Eugene Megnassan
Email: megnase@yahoo.com;
Tel.: +225-02-36-30-08 (F.L.)
© 2019 Sift Desk Journals. All Rights Reserved
VOLUME: 5 ISSUE: 1
Page No: 504-538
Eugene Megnassan
Email: megnase@yahoo.com;
Tel.: +225-02-36-30-08 (F.L.)
Issouf Fofana a, Brice Dali a,b, Frederica Mansilla-Koblavib, Eugene Megnassan a,b,c,d,f,*, Vladimir Frecer e,f and Stanislav Miertus f,g
a Laboratoire de Physique Fondamentale et Appliquée (LPFA), University of Abobo Adjamé (now Nangui Abrogoua), Abidjan, Côte d’Ivoire; fofetude@yahoo.fr, megnase@yahoo.com
b Laboratoire de Cristallographie – Physique Moléculaire, University of Cocody (now Felix Houphouët-Boigny), Abidjan, Côte d’Ivoire; fmkoblavi@hotmail.com
c Laboratoire de Chimie Organique Structurale et Théorique, University of Cocody (now Felix Houphouët-Boigny), Abidjan, Côte d’Ivoire;
d International Centre for Theoretical Physics, ICTP-UNESCO, Strada Costiera, Trieste, Italy;
e Department of Physical Chemistry of Drugs, Faculty of Pharmacy, Comenius University in Bratislava, Bratislava SK-83232, Slovakia; frecer@fpharm.uniba.sk
f International Centre for Applied Research and Sustainable Technology, Bratislava SK-84104, Slovakia; stanislav.miertus@icarst.org
g Department of Biotechnologies, Faculty of Natural Sciences, University of SS. Cyril and Methodius, Trnava SK-91701, Slovakia;
Issouf Fofana, Brice Dali, Frederica Mansilla-Koblavi, Eugene Megnassan, Vladimir Frecer, Stanislav Miertus, Structure-Based Design of Tetrahydroisoquinoline-Based Hydroxamic Acid Derivatives Inhibiting Human Histone Deacetylase 8(2021)Journal of Computational Chemistry & Molecular Modeling 5(1) : 504-538
We have designed new human histone deacetylase 8 (HDAC8) inhibitors using structure-based molecular design. 3D models of HDAC8–inhibitor complexes were prepared by in situ modification of the crystal structure of HDAC8 co-crystallized with the hydroxamic acid suberoylanilide (SAHA) and a training set (TS) of tetrahydroisoquinoline-based hydroxamic acid derivatives (DAHTs) with known inhibitory potencies. A QSAR model was elaborated for the TS yielding a linear correlation between the computed Gibbs free energies (GFE) of HDAC8–DAHTs complexation (∆∆Gcom) and observed half-maximal enzyme inhibitory concentrations (IC50exp). From this QSAR model a 3D-QSAR pharmacophore (PH4) was generated. Structural information derived from the 3D model and breakdown of computed HDAC8–DAHTs interaction energies up to individual active site residue contributions helped us to design new more potent HDAC8 inhibitors. We obtained a reasonable agreement ∆∆Gcom and values: (pIC50exp = – 0.0376 × ∆∆Gcom + 7.4605, R2 = 0.89). Similar agreement was established for the PH4 model (pIC50exp = 0.8769 × + 0.7854, R2 = 0.87). A comparative analysis of the contributions of active site residues guided the choice of fragments used in designing a virtual combinatorial library (VCL) of DAHT analogs. The VCL of more than 17 thousand DAHTs was screened by the PH4 and furnished 229 new DAHTs. The best-designed analog displayed predicted inhibitory potency up to 110 times higher than that of DAHT1 (IC50exp = 0.047 µM). Predicted pharmacokinetic profiles of the new analogs were compared to current per oral anticancer compounds. This computational approach, which combines molecular modelling, pharmacophore generation, analysis of HDAC8–DAHTs interaction energies and virtual screening of a combinatorial library of DAHTs resulted in a set of proposed new HDAC8 inhibitors. It can thus direct medicinal chemists in their search for new anticancer agents.
Cancer represents one of major public health problems. It remains a very common disease, in 2018, the cancer burden reached 18.1 million new cases and caused 9.6 million deaths worldwide [1]. Non communicable
Diseases (NCDs) are now responsible for the majority of deaths [1], and cancer is expected to be the leading cause of death and the single most important barrier to increasing life expectancy in all countries of the world in the 21st century[1]. There are several types of cancers whose origin can rest in genetic alterations or epigenetic deregulations. By altering the expression of genes involved in cellular regulation, epigenetic modifications, such as histone acetylation, play a fundamental role in the initiation and progression of tumours. Indeed, it has been shown that the breakdown of balance between acetylation and deacetylation levels of chromatin is involved in the acquisition of malignant phenotype [2] Overexpression of histones deacetylases (HDACs) induces low level of histone acetylation leading to silenced regulatory genes and in turn to human diseases such as escape of persistent HIV infection from latency [3], cancer [4,5,6,7,8,9, 10,11,12], as well as neurodegenerative, immune [5] and cardiac disorders [13].
Histones deacetylases are enzymes that catalyse the deacetylation of lysine residues located at the N-terminus of various protein substrates, such as histone nucleosomes. Hydrolysis of the acetyl group from histones results in condensed chromosomal DNA and transcriptional repression (figure 1) [14, 15, 16]. There are 18 human HDACs grouped into four classes [17, 18] according to sequence homology, function, DNA similarity, and phylogenetic analysis [19, 20, 21, 22] : classes I (HDACs 1-3 and 8), II (HDACs 4-7, 9 , and 10), and IV (HDAC 11) are zinc-dependent metallohydrolases, termed "classical HDACs," [18] while class III HDACs (sirtuins 1-7) are NAD+ dependent [23]. All zinc-dependent HDACs carry highly conserved catalytic site [24]. HDACs play a critical role in the regulation of many biological processes, including cell differentiation, proliferation, senescence, and apoptosis [25, 26]. HDACs are approved targets for drug design. Therefore, HDAC inhibition has become a common therapeutic strategy using inhibitors alone or in combinations. Indeed, inhibition of histone deacetylase results in growth arrest, differentiation, and apoptosis in almost all cancer cell lines, thus promoting HDACs as promising targets for antitumor therapy [25]. To date, four inhibitors of histone deacetylases (HDACi) have been approved by the Food & Drug Administration (FDA) for the treatment of cancers: hydroxamic acid suberoylanilide (SAHA, Vorinostat, Zolinza®), romidepsin (FK228, Istodax®), belinostat (Beleodaq®), and panobinostat (Farydak®). Other HDACi are currently in clinical trials including hydroxamic acids such as givinostat (ITF-2357), SB-939, R306465, and CRA024781, benzamides, such as entinostat (MS-275), mocetinostat (MGCD-0103), and tacedinaline (CI-994), and aliphatic acids, such as valproic acid, sodium phenylbutyrate, and pivanex (AN-9) [26, 27].
In this work, we design new analogues of tetrahydroisoquinoline-based hydroxamic acid derivatives (DAHT) from a series of 24 known DAHTs with specific experimental inhibition activities (IC50exp), which have been used as a training set of HDACi [25]. Tetrahydroisoquinoline-based hydroxamic acid derivatives is a series of inhibitors of histone deacetylases developed by Y. Zhang et al.[28] DAHT have been identified and validated as a potent histone deacetylase inhibitor (HDACi) with marked anti-tumor power in vitro and in vivo.[29] DAHTs are less toxic compared to pan-HDACi inhibitors [trichostatin A (TSA) and suberoylanilide hydroxamic acid (SAHA)] and could target other regulatory pathways to influence tumor metastases [30], such as aminopeptidase N(APN/CD13), also a Zn2+ dependent metalloprotease responsible for tumor invasion and angiogenesis [30]. The molecular structure of DAHTs plays an important role in the inhibition of HDAC or APN / CD13 compared to SAHA. [29] DAHTs exhibit excellent in vivo anticancer activities in a human breast carcinoma (MDA-MB-231), in a mouse hepatoma-22 (H22) pulmonary metastasis model and similar in vivo antitumor potency in a human colon tumor (HCT116) xenograft model and a potent growth inhibition in multiple tumor cell lines. [25, 30].
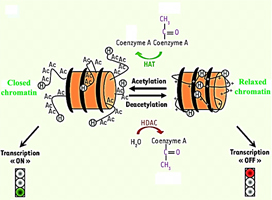
Figure 1. Schematic representation of DNA accessibility as a function of acetylation of the amino-terminal tails of histones.[3]
Tetrahydroisoquinoline bearing a hydroxamic acid is an excellent template to develop novel HDACi as potential anticancer agents.[25] From this training set, we have started to build an inhibition QSAR model of human histone deacetylase 8 (HDAC8) using a relevant descriptor [Gibbs free energy (GFE) of HDAC8-DAHT complex formation], by correlating computed GFE with experimental IC50exp. Each complex was carefully constructed by in situ modification of the reference crystal structure of HDAC8 (PDB entry 4QA0) [1]. Subsequently, a 3D-QSAR pharmacophore protocol was used to prepare four features pharmacophore model (PH4) from the bound conformations of DAHT inhibitors in the catalytic site of the HDAC8. The robustness of the PH4 model was based on the structural information of the HDAC8-DAHT complexes. The PH4 model was used to screen a virtual combinatorial library (VCL) of DAHTs with the goal to design more powerful bioavailable DAHT analogues inhibiting the HDAC8. The workflow describing the steps of the whole process of virtual design of novel DAHT analogues is presented in Scheme 1
The methodology of computer-assisted molecular design based on 3D models of E:I complexes and QSAR analysis of a known inhibitors training set has been successfully applied to optimization of antiviral, antibacterial, and anti-protozoan compounds including peptidomimetic, hydroxynaphthoic, thymidine, triclosan, pyrrolidine carboxamide derivatives, peptidic, ART hybrids, E64 epoxysuccinate and nitrile adazepeptide inhibitors [1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14].
2.1. Training and validation sets
Chemical structures and experimental biological activities (IC50exp) of training and validation sets of tetrahydroiso-quinoline-based hydroxamic acid derivatives inhibitors of HDAC8 used in this study were taken from literature [25]. The potencies of these compounds cover a sufficiently broad range of experimental half-maximal inhibitory concentrations (0.047 µM ≤ IC50exp ≤ 2.14 µM) to allow construction of a QSAR model. The training set (TS) containing 24 DAHT inhibitors and the validation set (VS) including 7 DAHT were taken from ref. [25]
2.2. Model building
Molecular modelling was carried out for the E:I (HDAC8:DAHT) complexes, free enzyme HDAC8, and free DAHT inhibitors starting from the high-resolution crystal structure of HDAC8 co-crystallized with the SAHA inhibitor (PDB entry code: 4QA0, resolution: 2.24 Å) using the Insight-II molecular modelling program [1].
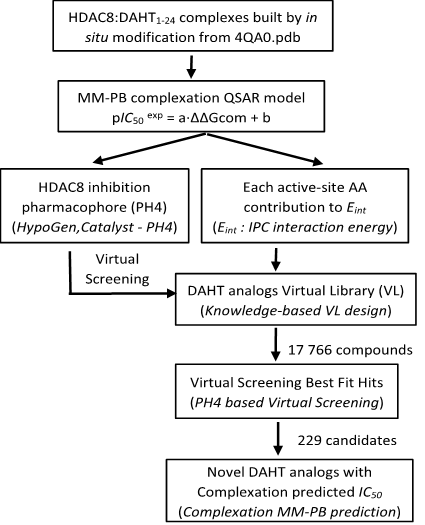
Scheme 1. Workflow describing the multistep approach to virtually design novel DAHT analogues with higher predicted potency against HDAC8.
Inhibitors were modelled from the 4QA0 reference crystal structure [32] by in situ modification of the molecular scaffold and function groups of the co-crystallized inhibitor SAHA. All rotatable bonds of the replacing fragments were subjected to an exhaustive conformational search coupled with a careful gradual energy-minimisation of the modified ligand and HDAC8 active site residues in the immediate vicinity (5Å radius) in order to identify low-energy bound conformations of the modified inhibitor. The resulting low-energy structures of the HDAC8:DAHT complexes were then carefully refined by energy-minimization procedure of the entire complex to obtain stable structures. The full description of the calculation of the ligand binding relative affinity (ΔΔGcom) is described in the reference [34].
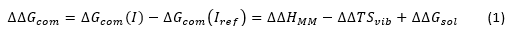
The ∆∆HMM describes the relative enthalpic contribution to the GFE change corresponding to the intermolecular interactions in the E:I complex estimated by molecular mechanics (MM). The ∆∆Gsol and ∆∆TSvib represent the relative solvation and vibrational entropy contributions to the GFE of the E:I complex formation, respectively.
2.3. Molecular mechanics
Modelling of the inhibitors and their complexes was carried out in the all-atom representation using atomic, bond and charges parameters of the Class II Consistent Force Field (CFF91) [33]. A dielectric constant of 4 was used for all MM calculations in order to take into account the dielectric shielding effect in proteins. Minimizations of the E:I complexes, free E and I were carried out by relaxing the structures gradually, starting with added hydrogen atoms, continued with residue side chain heavy atoms and followed by the protein backbone relaxation. Geometry optimizations were performed using an enough steepest descent and conjugate gradient iterative cycles and average gradient convergence criterion of 0.01 kcal.mol-1.Å-1.
2.4. Conformational search
Free inhibitor conformations were derived from their bound conformations in the E-I complexes by gradual relaxation to the nearest local energy minimum as described earlier34.
2.5. Solvation Gibbs free energies
The electrostatic component of the solvation GFE, which includes also the effect of ionic strength of the solvent by solving the nonlinear Poisson–Boltzmann equation [[i],[ii]] was computed by the DelPhi module of the Discovery Studio (DS 2.5) [[iii]]. The program represents the solvent by a continuous medium of high dielectric constant (εro = 80) and the solute as a charge distribution filling a cavity of low dielectric (εri = 4) with boundaries linked to the solute’s molecular surface. The program numerically solves for the molecular electrostatic potential and reaction field around the solute using finite difference method. DelPhi calculations were done on a (235 x 235 x 235) cubic lattice grid for the E:I complexes and free E and on a (65 x 65 x 65) grid for the free I. Full coulombic boundary conditions were employed. Two subsequent focusing steps led to a similar final resolution of about 0.3 Å per grid unit at 70% filling of the grid by the solute. Physiological ionic strength of 0.145 mol.dm-3, atomic partial charges and radii defined in the CFF91 force field parameter set [50] and a probe sphere radius of 1.4 Å were used. The electrostatic component of the Poisson–Boltzmann solvation GFE was calculated as the reaction field energy [41, 43, 48, 49, 4, 5, 6].
2.6. Calculation of binding affinity and QSAR model
The calculation of binding affinity expressed as complexation GFE has been described fully earlier [34].
2.7. Interaction energy
The molecular mechanics interaction energy (Eint) calculation protocol available in Discovery Studio 2.5 [50] was used to compute the non-bonded interactions (van der Walls and electrostatic interatomic potential terms) between two sets of atoms belonging either to the E or I in the E:I complexes. All pairs of interactions of the total enzyme-inhibitor interaction energy were evaluated using CFF91 force field parameters with a relative permittivity of 4 [50]. In particular, the breakdown of Eint into contributions from individual active site residues allows a quantitative analysis, which permits identification of residues with the highest contribution to the ligand binding. It also helps with rapid identification of favourable structural modifications and suggests molecular moieties in the inhibitor structure which are primarily responsible for receptor binding and biological activity of the compound33.
2.8. Pharmacophore generation
Bound conformations of inhibitors taken from the models of E:I complexes were used for building of 3D QSAR pharmacophore by means of the HypoGen algorithm of Catalyst [1] implemented in Discovery Studio [50]. The top scoring pharmacophore hypothesis was built up in three steps (constructive, subtractive, and optimization steps) from the set of most active inhibitors. Inactive molecules served for definition of the excluded volume. The maximum number of five features allowed by the HypoGen algorithm was selected based on the DAHT scaffold and substituents during the pharmacophore generation, namely: positive ionizable (POS_IONIZABLE), hydrophobic aliphatic (HYd), hydrogen bond donor, (HBD), hydrogen bond acceptor (HBA), and ring aromatic (Ar). Adjustable parameters of the protocol were kept at their default values except the uncertainty on the activity, which was set to 1.25 instead of 3.This last choice to bring the uncertainty interval on experimental activity from the large[IC50exp/3; 3x IC50exp] to a relatively narrow[4 x IC50exp /5; 5 x IC50exp /4], due to accuracy and homogeneity of the measured activities originating from the same laboratory [25]. During the generation of 10 pharmacophores, the number of missing features was set to 0 and the best one was selected. Generally, a PH4 model, as the one described here, can be used to estimate predicted activities of new analogues based on their mapping to the PH4 features. In this study, priority was given to PH4 screening of VCL of DAHT analogues.
2.9. ADME properties
Properties that determine the pharmacokinetics profile of a compound, besides octanol/water partitioning coefficient, aqueous solubility, blood/brain partition coefficient, Caco-2 cell permeability, serum protein binding, number of likely metabolic reactions and other 18 descriptors related to adsorption, distribution, metabolism and excretion (ADME properties) of the inhibitors were computed by the QikProp program [1] based on the methods of Jorgensen [2,3,4]. According to these methods, experimental results of more than 710 compounds including about 500 drugs and related heterocycles were correlated with computed physicochemical descriptors, resulting in an accurate prediction of molecular pharmacokinetic profiles. Drug likeness (#stars) is represented by the number of descriptors that exceed the range of values determined for 95% of known drugs out of 24 selected descriptors computed by the QikProp[55]. Drug-likeness was used as the global compound selection criterion related to ADME properties. The selected ADME descriptors were calculated from 3D structures of the DAHTs considered. They were used to assess the pharmacokinetics profile of designed compounds and served also for the VCL focusing.
2.10. Virtual library generation
The analogue model building was performed with Discovery Studio 2.5 [50]. The library of analogues was enumerated by attaching R-groups (fragments, building blocks) onto DAHT scaffold using the Enumerate Library from Ligands module of Discovery Studio 2.5 [50]. Reagents and chemicals considered in this paper were selected from the catalogues of chemicals available from the commercial sources. Each analogue was built as a neutral molecule, its geometry was refined by MM optimization through smart minimizer of Discovery Studio 2.5 [50] meeting high convergence criteria (threshold on energy difference of 10-4 kcal.mol-1 and root mean square deviation (RMSD) of 10-5 Å), dielectric constant of 4, using class II consistent force field CFF91 [33].
2.11. ADME-based library searching
Twenty four ADME-related molecular descriptors available in QikProp [55], which characterize a wide spectrum of molecular properties as described in Section 4.9, were used. Optimum ranges of these 24 descriptors were defined in terms of upper and lower bounds according to QikProp [55]. Among them predicted drug-likeness (#stars, Section 4.9) was used to retain drug-like DAHT analogues in the focused VCL.
2.12. Pharmacophore-based library searching
The pharmacophore model (PH4) described in Section 4.8 was derived from the bound conformations of DAHTs at the active site of human HDAC8. The enumerated VCL was screened by pharmacophore mapping protocol available of Discovery Studio [50]. Within this protocol, each generated conformer of the analogues was geometry optimized by means of the CFF91 force field for a maximum of 500 energy minimization steps and subsequently aligned and mapped to the PH4 model in order to select the top-ranking overlaps. Twenty best-fitting inhibitor conformers were saved and clustered into 10 conformational families according to their mutual RMSD by Jarvis-Patrick complete linkage clustering method [1]. The best representative of each cluster was retained for the virtual screening of analogues. Only those analogues mapping to all PH4 features were retained for the in silico screening.
2.13. Inhibitory potency prediction
The conformer with the best match to the PH4 pharmacophore in each cluster of the focused library subset was selected for in silico screening by the complexation QSAR model. The relative GFE of E:I complex formation in water ∆∆Gcom was computed for each selected new analogue and then used for prediction of HDAC8 inhibitory potencies (IC50pre) of the focused VCL of DAHT analogues by inserting this parameter into the target-specific scoring function. The scoring function, which is specific for the HDAC8 receptor, given in equation (2), was parameterized using the QSAR model of the training set of DAHT inhibitors [25].
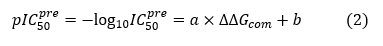
3.1. Training and validation sets
A series of 31 [24 training set (TS) DAHTs and 7 validation set (VS) DAHTs] of DAHT inhibitors and their experimental activities (IC50exp) from the same laboratory [25] were selected (Table 1). These cover a relatively wide range of potencies 0.047 μM ≤ IC50exp ≤ 2.14 μM and allowed building of a valid QSAR model. The chirality label (when applicable) of the inhibitor atoms is displayed in the last part of Table 1 on the molecular structure of each TS and VS inhibitor [25].
3.2. QSAR model
3.2.1. One descriptor QSAR model
The relative Gibbs free energy (GFE) of E:I complex formation ΔΔGcom was calculated for the HDAC8:DAHT complexes as described in Section 3. Table 2 shows the GFE and their components, equation (1). The ΔΔGcom reflects the mutual binding affinity between the enzyme and the inhibitor. Since it is calculated via an approximate approach, the relevance of the binding model is evaluated by a linear regression with experimentally observed activity data (IC50exp) [25], equation (2), which led to a linear correlation and QSAR model for the training set of DAHT inhibitors. One correlation equation obtained for the GFE of E:I complex formation ΔΔGcom (equation (A)), presented in Table 3 with the relevant statistical data. The relatively high values of the regression coefficient R2 and the Fischer F-test of the correlation involving ΔΔGcom indicate that there is a strong relationship between the binding model and the experimental inhibitory potencies of the DAHT. The statistical data confirmed validity of the correlation equation (A) plotted on Figure 2. The ratio pIC50pre / pIC50exp ≅ 1(the pIC50pre values were estimated using correlation eq. A, Table 3.) calculated for the validation set DAHT25-31 documents the substantial predictive power of the complexation QSAR model from Table 2. Thus, the regression equation A (Table 3) and computed ΔΔGcom GFEs can be used for prediction of inhibitory potencies against human HDAC8 for novel DAHT analogues, provided that they share the same binding mode as the training set tetrahydroisoquinoline-based hydroxamic acid derivatives DAHT1-24.
Table 1. Training set (TS) and validation set (VS) of DAHT inhibitors [25] of human HDAC8 used in the preparation of QSAR model of HDAC8 inhibition. The last part of the table represents molecular structure of TS and VS indicating the chirality label (when applicable) of the inhibitor atoms [25].
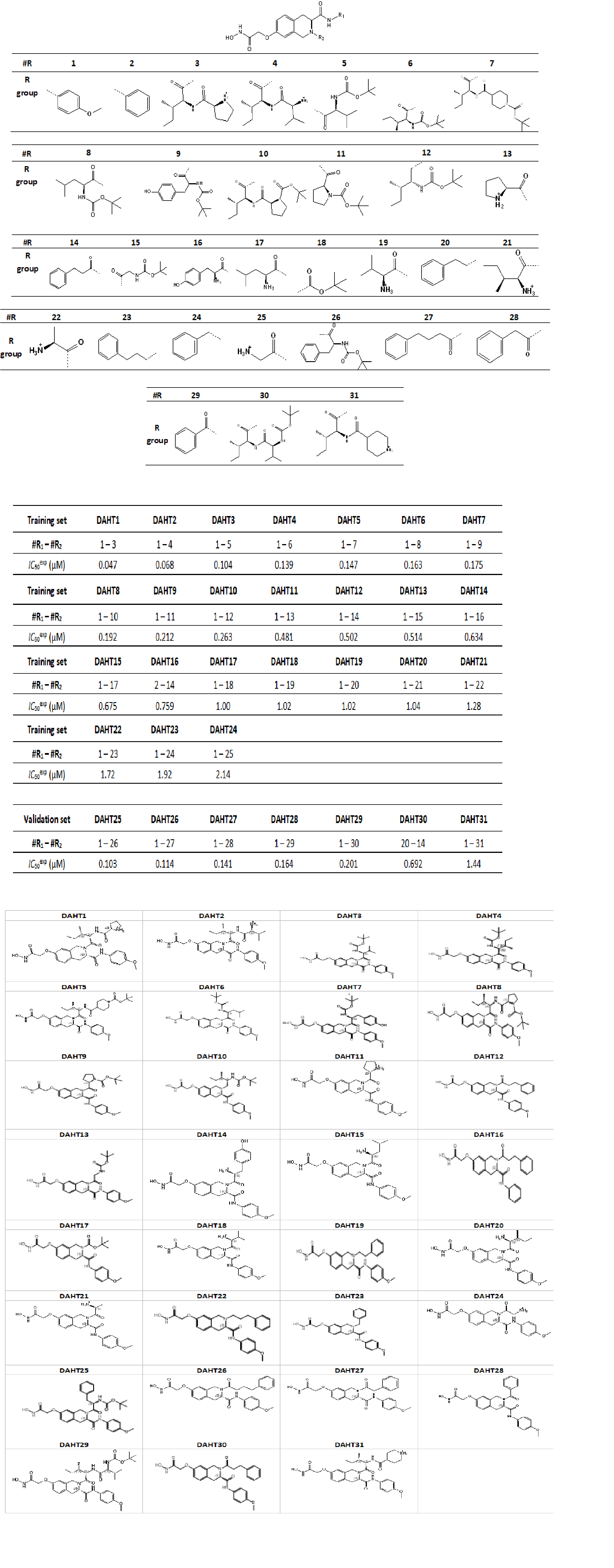
Table 2 Complexation Gibbs free energy (binding affinity) and its components for the training set of human HDAC8 inhibitors DAHT1-24 and validation set inhibitors DAHT25-31 [25]
|
Training set a |
Mw b |
∆∆HMM c |
∆∆Gsol d |
ΔΔTSvib e |
∆∆Gcom f |
IC50expg |
|
[g×mol-1] |
[kcal×mol-1] |
[kcal×mol-1] |
[kcal×mol-1] |
[kcal×mol-1] |
[µM] |
|
|
DAHT1 |
583 |
0.00 |
0.00 |
0.00 |
0.00 |
0.047 |
|
DAHT2 |
585 |
19.96 |
-5.10 |
2.69 |
12.16 |
0.068 |
|
DAHT3 |
571 |
69.08 |
-46.89 |
2.57 |
19.62 |
0.104 |
|
DAHT4 |
585 |
67.15 |
-46.61 |
2.85 |
17.70 |
0.139 |
|
DAHT5 |
696 |
65.59 |
-43.39 |
5.28 |
16.92 |
0.147 |
|
DAHT6 |
585 |
68.81 |
-46.83 |
2.93 |
19.05 |
0.163 |
|
DAHT7 |
635 |
67.39 |
-43.95 |
-4.43 |
27.87 |
0.175 |
|
DAHT8 |
682 |
64.23 |
-44.67 |
1.81 |
17.75 |
0.192 |
|
DAHT9 |
541 |
72.18 |
-48.39 |
-0.87 |
24.66 |
0.212 |
|
DAHT10 |
462 |
70.42 |
-46.04 |
4.02 |
20.36 |
0.263 |
|
DAHT11 |
470 |
32.32 |
-2.71 |
-5.82 |
35.43 |
0.481 |
|
DAHT12 |
504 |
72.23 |
-48.02 |
-7.21 |
31.42 |
0.502 |
|
DAHT13 |
529 |
70.65 |
-46.15 |
-3.74 |
28.24 |
0.514 |
|
DAHT14 |
535 |
25.07 |
0.42 |
-6.35 |
31.83 |
0.634 |
|
DAHT15 |
486 |
24.08 |
2.70 |
-6.74 |
33.53 |
0.675 |
|
DAHT16 |
474 |
73.48 |
-47.89 |
-9.13 |
34.72 |
0.759 |
|
DAHT17 |
472 |
74.32 |
-47.77 |
-5.20 |
31.76 |
1.00 |
|
DAHT18 |
472 |
28.68 |
-1.16 |
-4.62 |
32.14 |
1.02 |
|
DAHT19 |
502 |
81.74 |
-46.76 |
-6.79 |
41.78 |
1.02 |
|
DAHT20 |
486 |
24.34 |
3.76 |
-3.44 |
31.54 |
1.04 |
|
DAHT21 |
443 |
31.20 |
0.27 |
-8.57 |
40.04 |
1.28 |
|
DAHT22 |
490 |
84.66 |
-46.68 |
-9.37 |
47.36 |
1.72 |
|
DAHT23 |
571 |
86.35 |
-44.82 |
-7.69 |
49.22 |
1.92 |
|
DAHT24 |
429 |
41.24 |
-2.68 |
-7.50 |
46.07 |
2.14 |
|
Validation set |
Mw b |
∆∆HMM c |
∆∆Gsol d |
ΔΔTSvib e |
∆∆Gcom f |
pIC50pre / pIC50exp h |
|
[g×mol-1] |
[kcal×mol-1] |
[kcal×mol-1] |
[kcal×mol-1] |
[kcal×mol-1] |
||
|
DAHT25 |
619 |
84.77 |
-44.95 |
-1.17 |
40.99 |
0.85 |
|
DAHT26 |
518 |
72.55 |
-45.75 |
-7.36 |
34.16 |
0.89 |
|
DAHT27 |
490 |
73.68 |
-47.35 |
-9.51 |
35.83 |
0.89 |
|
DAHT28 |
476 |
74.84 |
-47.07 |
-11.07 |
38.83 |
0.88 |
|
DAHT29 |
684 |
66.21 |
-45.88 |
8.29 |
12.04 |
1.05 |
|
DAHT30 |
476 |
68.06 |
-47.63 |
-7.27 |
27.70 |
1.04 |
|
DAHT31 |
597 |
21.18 |
-1.93 |
1.12 |
18.14 |
1.16 |
a for the chemical structures of the training and validation sets of inhibitors see Table 1;
b Mw is the molecular mass of inhibitors;
c DDHMM is the relative enthalpic contribution to the GFE change related to E-I complex formation derived by MM:
DDHMM ⋍ [EMM{E-Ix} - EMM{Ix}] - [EMM{E-Iref} - EMM{Iref}], Iref is the reference inhibitor DAHT1;
d DDGsol is the relative solvent effect contribution to the GFE change of E-I complex formation:
DDGsol = [Gsol{E-Ix} - Gsol{Ix}] - [Gsol{E-Iref} - Gsol{Iref}];
e -DDTSvib is the relative entropic contribution of inhibitor Ix to the GFE of E-Ix complex formation:
DDTSvib = [TSvib{Ix}E - TSvib{Ix}] - [TSvib{Iref}E - TSvib{Iref}];
f DDGcom is the overall relative GFE change of E-Ix complex formation: DDGcom ⋍ DDHMM + DDGsol - DDTSvib;
g IC50exp is the experimental half-maximal inhibition concentration of DAHT obtained from ref.[25];
h ratio of predicted and experimental half-maximal inhibition concentrations pIC50pre / pIC50exp (pIC50pre = -log10 IC50pre) was
predicted from computed DDGcom using the regression equation for DAHT shown in Table 3, A.
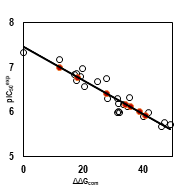
Figure 2. Plot for relative complexation Gibbs free energies (GFE) of the HDAC8-DAHTx complex formation ∆∆Gcom [kcal×mol-1] of the training set [25]. The validation set data points are shown in red colour.
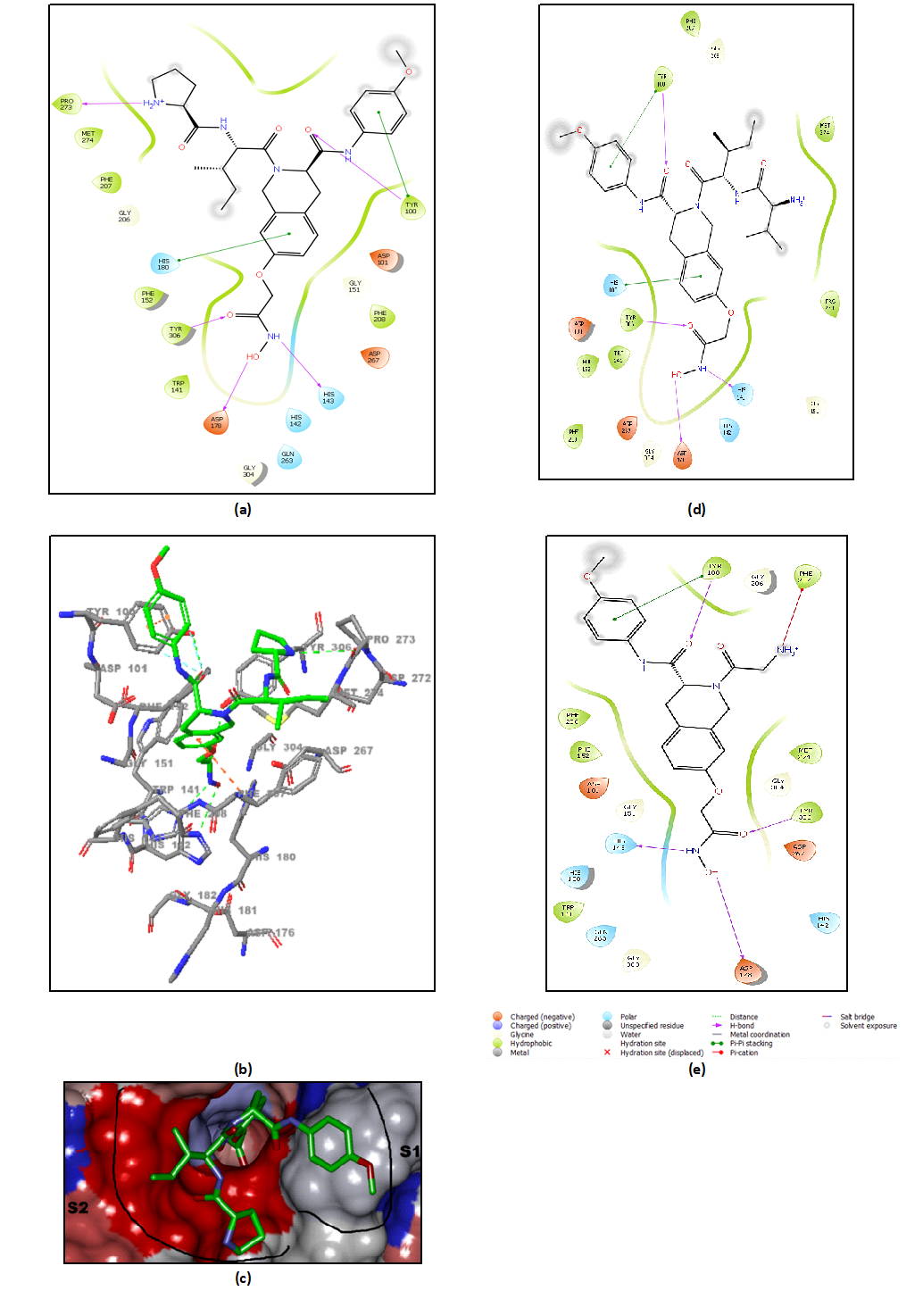
Figure 3. (a) - 2D schematic interaction diagram of the most potent inhibitor DAHT1[25] at the active site of human HDAC8. (b) - 3D structure of the HDAC8 active site with bound inhibitor DAHT1. (c) - Connolly surface of the HDAC8 active site for DAHT1. Surface colouring legend: red - hydrophobic, blue - hydrophilic and white - intermediate. (d) - 2D schematic interaction diagram of the inhibitor DAHT2 [25] at the active site of human HDAC8. (e) - 2D schematic interaction diagram of the inhibitor DAHT24 [25] at the active site of human HDAC8.
Table 3. Regression analysis of computed binding affinities ∆∆Gcom, its enthalpic component ∆∆HMM, and experimental half-maximal inhibitory concentrations pIC50exp = -log10 IC50exp of DAHTs towards HDAC8.
|
Statistical data of linear regression |
(A) |
|
|
pIC50exp -0.0376×∆∆Gcom+7.4605 (A) |
|
|
|
Number of compounds n |
24 |
|
|
Squared correlation coefficient of regression R2 |
0.89 |
|
|
LOO cross-validated squared correlation coefficient R2xv |
0.88 |
|
|
Standard error of regression s |
0.163 |
|
|
Statistical significance of regression, Fisher F-test |
171.58 |
|
|
Level of statistical significance a |
> 95% |
|
|
Range of activities IC50exp [µM] |
0.047-2.14 |
|
3.2.2. Binding mode of DAHTs
Structural information on the enzyme-inhibitor interactions retrieved from the crystal structure of HDAC8-DAHT1-2 complexes [25] showed that DAHTs are micromolar HDAC8 inhibitors. As indicated in Figure 3, in the catalytic site I residue Tyr100 forms p - p stacking interaction [1] with the 4-methoxyphenyl group of inhibitor [25] and a hydrogen bond (HB) with carbonyl oxygen atom of DAHT scaffold. The central benzene ring of the inhibitor scaffold forms p - p stacking interaction with His180. In the hydrophobic site II, the ((1S, 2R) -2-methyl-1 - ((R) -pyrrolidine-2-carboxamido) butyl) moiety of DAHT1 (figure 3.a) and ((1S, 2R) -1 - ((S) -2- amino-3-methylbutanamido) -2-methylbutyl) moiety of DAHT2 (figure 3.d) sit in a hydrophobic substrate cavity, surrounded by side chains of predominantly nonpolar residues: Phe207, Pro273 and Met274. In the case of the inhibitor DAHT24, the aminomethyl moiety (figure 3.e) is shorter, less bulky and cannot reach this aforementioned cavity therefore interacts weakly with the residue Phe207 and even less the residues Pro273 and Met274. We postulated that the high affinity between the DAHT1 R2 group and these three residues, shown by the pocket interaction energy, could be the key factor that made DAHT1 more effective against HDAC8 compared to another training set inhibitors. In the hydrophilic site III, the zinc-binding group (ZBG) of inhibitor scaffold makes hydrogen bonds with the side chain of catalytic His143, Asp178, and Tyr306.
3.3. Interaction Energy
Other key structural information was provided by the interaction energy (IE, ΔEint) diagram obtained for each training set inhibitor. IE breakdown to contributions from HDAC8 active site residues is helpful for the choice of relevant R1-groups (site I) and R2-groups (site II), which could improve the binding affinity of DAHT analogues to the human HDAC8 and the subsequently enhance the inhibitory potency. A comparative analysis of computed IE for training set DAHTs (Figure 4) divided into three classes according to the range of experimental activities (0.047-2.14µM) of training set DAHTs (highest, moderate and lowest activity) has been carried out to identify the residues for which the interaction with the ligand could be increased. However, the comparative analysis showed about the same level of IE contributions from site I residues for all three classes of inhibitors, which seems normal to us since it is the 4-methoxyphenyl group that is used in all the training set inhibitors in this pocket. Only inhibitor DAHT16 contains phenyl group in R1 which results in a decrease in the contribution of Tyr100 to the IE. (Le DAHT16 n’est pas représenté sur le diagramme IE) Contrariwise, we observed a decrease in the IE contributions of the residues Lys202, Phe207, Pro273, Met274 and the cofactor UNK405H from site II of class (A) to class (C). Therefore, we have adopted a combinatorial approach to novel analogue design with help of the PH4 pharmacophore of HDAC8 inhibition derived from the complexation QSAR model. Starting from the best combinatorically designed analogue, we proceeded by the method of intuitive substitution allowing to improve the binding affinity as we previously reported for the thymine-like inhibitors of Mycobacterium tuberculosis thymidine monophosphate kinase design [33].
3.4. 3D-QSAR Pharmacophore model
3.4.1. HDAC8 active site pharmacophore
The Connolly surface generation protocol in Insight-II molecular modelling program [47] allows for mapping of hydrophobic and hydrophilic character of the active site of a protein. The surface of the active site of HDAC8 has mainly a hydrophobic character (Figure 3, c).
3.4.2. Generation and validation of 3D-QSAR pharmacophore
HDAC8 inhibition 3D-QSAR pharmacophore was generated from the active conformation of 24 TS inhibitors DAHT1-24 and evaluated by 7 VS DAHT25-31 with the range of experimental activities (0.047 – 2.14 µM) spanning more than two orders of magnitude [25]. The PH4 pharmacophore model of the HDAC8 inhibition elaborated from QSAR model and training set of DAHT is presented on Figure 5. The 3D-QSAR PH4 generation was carried out in three steps: constructive, subtractive and optimization step (Section 2). During the constructive phase of HypoGen, the most active DAHTs, for which IC50exp ≤ 2 x 0.047 μM, were selected as the leads. Thus, DAHT1 and DAHT2 (IC50exp ≤ 0.094 μM) were used to generate the starting PH4 features and those matching to these leads were retained. During the subsequent subtractive phase, features which were present in more than half of the inactive DAHTs were removed. The PH4 models that contained all features were retained. None of the training set compounds was found to be inactive (IC50exp > 0.047 x 103.5 = 148.63 μM). During the final optimization phase, the score of the PH4 hypothesis was improved. Hypotheses were scored via simulated annealing protocol according to errors in the activity estimates from the regression and complexity. At the end of optimization, 10 best scoring unique hypotheses (Table 4) displaying four features were kept.
The reliability of the generated PH4 models was then assessed using the calculated cost parameters ranging from 98.68 (Hypo1) to 130.99 (Hypo10). Their statistical data (costs, root-mean-square deviation 1.75 ≤ RMSD ≤ 2.32 and 0.87 ≤ ≤ 0.93 are listed in Table 4. The regression equation: pIC50exp = 0.8769 x IC50pre + 0.7854 (Figure 5), both and greater than 0.85 as well as F-test of 143.51 attest the predictive capacity of the PH4. The fixed cost of Hypo1 (57.18), lower than the null cost (318.83) by Δ = 261.65, is a chief indicator of the PH4 model predictability (Δ > 70 corresponds to a probability higher than 90% that the model represents a valid correlation [33]). The difference Δ ≥ 187.8 for the set of 10 hypothesis confirms the high quality of the PH4 model. The best-selected hypothesis Hypo1 represents a PH4 model with a similar level of predictive power as the QSAR model utilizing the GFE of E:I complex formation with a probability of 98%.
The substantial predictive power of the generated PH4 model was also checked through the computed ratio of PH4-predicted and experimentally observed activities (pIC50pre / IC50exp) for the validation set (VS) (Table 1). All these ratios computed for the DAHT25-DAHT31 are close to one, Table 2.
3.5. Virtual Screening
In silico screening of virtual library of ligands can lead to hit identification as it was shown in our previous works on inhibitor design [34,35,36].
3.5.1. Virtual library
An initial virtual combinatorial library (VCL) was generated by substitutions at positions R1 and R2 (see Table 1) on the tetrahydroisoquinoline-based hydroxamic acid derivatives scaffold. During the VCL enumeration, the R-groups listed in Table 5 were attached to positions R1 – R2 of the DAHT scaffolds to form a virtual combinatorial library of the size: R1 x R2 = 42 x 423 = 17 766 analogues (Table 5). In order to match the substitution pattern of the best training set inhibitor DAHT1 and taking into account the reported structural information about the character of the binding site pockets [25], without applying the Lipinski rules [1], the VCL underwent focusing.
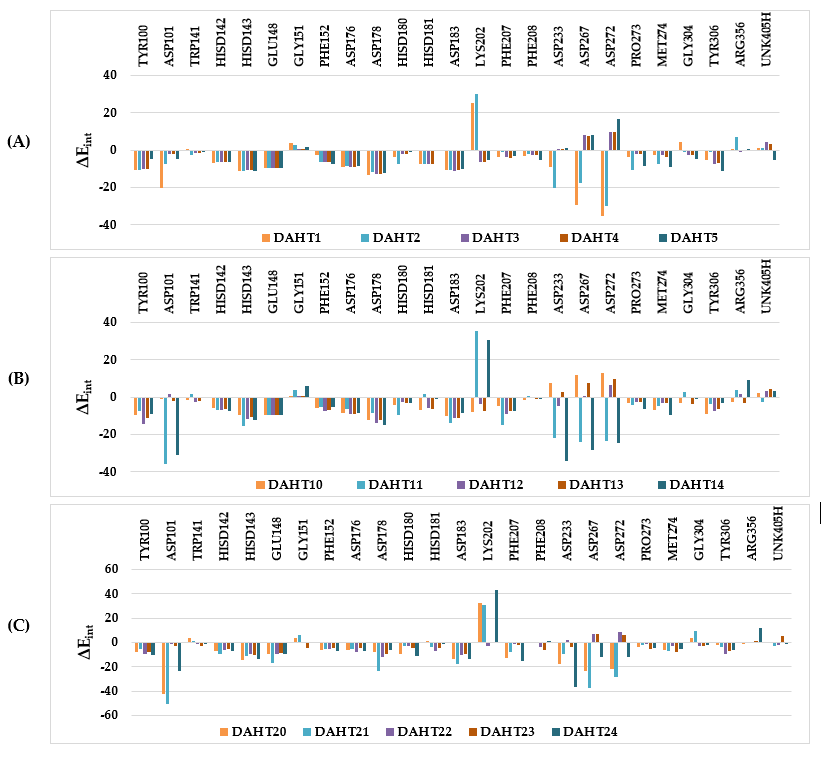
Figure 4 : Molecular mechanics intermolecular interaction energy ΔEint breakdown to residue contributions in [kcal.mol-1]: (A) - the most active inhibitors DAHT1-5, (B) - moderately active inhibitors DAHT 10-14, (C) - less active inhibitors DAHT20-24, Table 2 [25].
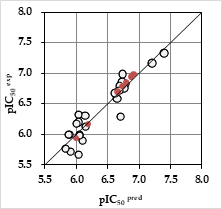
Figure 5. The correlation plot of experimental vs. predicted inhibitory activity (open circles correspond to TS, orange dots to VS).
Table 4. Output parameters of 10 generated pharmacophore hypotheses for training set of DAHT HDAC8 inhibitors [25] after the CatScramble validation procedure.
|
Hypothesis |
RMSD a |
R2 b |
Total costs c |
Costs Difference d |
Closest Random e |
|
Hypo1 |
1.750 |
0.93 |
98.68 |
220.15 |
150.84 |
|
Hypo2 |
1.936 |
0.92 |
108.51 |
210.33 |
170.91 |
|
Hypo3 |
1.978 |
0.91 |
111.64 |
207.19 |
171.52 |
|
Hypo4 |
2.048 |
0.90 |
115.31 |
203.53 |
189.92 |
|
Hypo5 |
2.064 |
0.90 |
115.80 |
203.04 |
194.45 |
|
Hypo6 |
2.113 |
0.90 |
117.87 |
200.97 |
197.90 |
|
Hypo7 |
2.320 |
0.88 |
128.49 |
190.34 |
200.00 |
|
Hypo8 |
2.358 |
0.87 |
130.18 |
188.65 |
201.67 |
|
Hypo9 |
2.328 |
0.87 |
130.64 |
188.20 |
208.89 |
|
Hypo10 |
2.320 |
0.88 |
130.99 |
187.84 |
209.65 |
a root mean square deviation; b squared correlation coefficient; c overall cost parameter of the PH4 pharmacophore;
d cost difference between null cost and hypothesis total cost, e lowest cost from 49 scrambled runs at a selected level of confidence of 98%. The Fixed cost = 57.18 with RMSD = 0, the Null cost = 318.83 with RMSD = 4.799 and the Configuration cost = 14.33.
3.5.2. In silico screening of library of DAHTs
The focused library of 17 766 analogues was further screened for molecular structures matching the 3D-QSAR PH4 pharmacophore model Hypo1 of HDAC8 inhibition. 199 DAHTs mapped to at least 3 pharmacophoric features, 30 of which mapped to at least 4 features of the pharmacophore. These best fitting analogues (PH4 hits) then underwent complexation QSAR model screening. The computed GFE of HDAC8-DAHTx complex formation, their components and predicted half-maximal inhibitory concentrations calculated from the correlation equation (A) (Table 3), are listed in Table 6.
Table 5. R1-R2-groups (building blocks) used in the design of the initial diversity virtual combinatorial library of tetrahydroisoquinoline-based hydroxamic acid derivatives
https://www.siftdesk.org/articles/images/10708/t5.pdf
Table 6. Complexation GFE and their components for the top-scoring 229 virtual DAHT analogues. The analogue numbering concatenates the index of each substituent R1 to R2 with the substituent numbers taken from Table 5.
|
N° |
Designed analogues R1 - R2 |
ΔΔHMM [kcal.mol-1] |
ΔΔGsol [kcal.mol-1] |
ΔΔTSvib [kcal.mol-1] |
ΔΔGcom [kcal.mol-1] |
IC50pre[nM] |
|
|
DAHT1 |
0 |
0 |
0 |
0 |
47 |
|
1 |
11-213 |
73.43 |
-45.79 |
0.06 |
27.60 |
378 |
|
2 |
10-214 |
83.97 |
-48.60 |
1.21 |
34.18 |
668 |
|
3 |
9-215 |
70.45 |
-47.09 |
-4.69 |
28.07 |
393 |
|
4 |
12-216 |
62.74 |
-44.07 |
1.99 |
16.70 |
147 |
|
5 |
13-217 |
64.38 |
-39.79 |
2.73 |
21.88 |
230 |
|
6 |
9-218 |
71.11 |
-44.61 |
-7.43 |
33.95 |
654 |
|
7 |
21-226 |
53.24 |
-39.22 |
-1.19 |
15.23 |
129 |
|
8 |
21-227 |
75.68 |
-46.79 |
2.25 |
26.66 |
348 |
|
9 |
16-222 |
61.57 |
-42.70 |
-7.98 |
26.87 |
355 |
|
10 |
17-223 |
64.60 |
-45.77 |
1.39 |
17.46 |
157 |
|
11 |
18-224 |
68.16 |
-41.88 |
6.10 |
20.20 |
199 |
|
12 |
19-20 |
66.67 |
-44.58 |
-3.50 |
25.61 |
318 |
|
13 |
20-225 |
30.87 |
-25.97 |
-5.90 |
10.82 |
88 |
|
14 |
1-219 |
68.51 |
-45.44 |
5.98 |
17.11 |
152 |
|
15 |
14-212 |
73.70 |
-47.66 |
-3.36 |
29.42 |
442 |
|
16 |
15-220 |
69.20 |
-39.81 |
-2.12 |
31.53 |
531 |
|
17 |
4-221 |
66.72 |
-44.50 |
-2.48 |
24.72 |
294 |
|
18 |
1-188 |
65.21 |
-45.37 |
-0.93 |
20.79 |
209 |
|
19 |
1-65 |
59.16 |
-43.59 |
2.60 |
12.99 |
107 |
|
20 |
1-246 |
53.09 |
-33.85 |
0.49 |
18.77 |
176 |
|
21 |
1-247 |
51.29 |
-45.04 |
4.96 |
1.31 |
39 |
|
22 |
1-248 |
51.97 |
-44.34 |
2.77 |
4.88 |
53 |
|
23 |
1-249 |
53.20 |
-42.75 |
-0.58 |
11.05 |
90 |
|
24 |
4-250 |
51.48 |
-43.45 |
-12.49 |
20.54 |
205 |
|
25 |
4-251 |
50.05 |
-39.93 |
-8.92 |
19.06 |
180 |
|
26 |
1-252 |
50.16 |
-42.76 |
-4.19 |
11.61 |
95 |
|
27 |
1-253 |
57.67 |
-43.20 |
-4.51 |
19.00 |
179 |
|
28 |
3-254 |
50.87 |
-40.85 |
-4.76 |
14.80 |
125 |
|
29 |
1-255 |
53.81 |
-36.58 |
1.49 |
15.76 |
135 |
|
30 |
1-256 |
50.16 |
-46.23 |
0.08 |
3.87 |
48 |
|
31 |
22-293 |
49.17 |
-33.43 |
1.43 |
14.33 |
120 |
|
32 |
4-277 |
-57.43 |
77.02 |
5.19 |
14.42 |
121 |
|
33 |
4-278 |
33.31 |
-24.34 |
3.52 |
5.47 |
56 |
|
34 |
4-279 |
28.91 |
-23.72 |
3.91 |
1.30 |
39 |
|
35 |
4-280 |
30.74 |
-24.91 |
2.24 |
3.61 |
47 |
|
36 |
4-281 |
42.35 |
-23.52 |
7.94 |
10.91 |
89 |
|
37 |
1-282 |
65.44 |
-43.56 |
10.01 |
11.89 |
97 |
|
38 |
257-283 |
54.74 |
-31.52 |
14.21 |
9.03 |
76 |
|
39 |
261-285 |
70.34 |
-42.21 |
11.56 |
16.59 |
146 |
|
40 |
261-286 |
72.36 |
-47.59 |
8.27 |
16.52 |
145 |
|
41 |
259-285 |
70.30 |
-39.87 |
7.87 |
22.58 |
245 |
|
42 |
260-285 |
74.65 |
-41.17 |
4.13 |
29.37 |
440 |
|
43 |
22-287 |
54.34 |
-24.80 |
-0.52 |
30.08 |
468 |
|
44 |
262-287 |
41.86 |
-24.58 |
-1.78 |
19.08 |
181 |
|
45 |
258-284 |
65.39 |
-46.95 |
1.13 |
17.33 |
155 |
|
46 |
1-288 |
46.31 |
-41.62 |
5.13 |
-0.42 |
33 |
|
47 |
263-289 |
58.87 |
-45.78 |
3.28 |
9.83 |
81 |
|
48 |
1-290 |
50.13 |
-40.38 |
7.33 |
2.44 |
43 |
|
49 |
264-290 |
47.56 |
-45.72 |
10.03 |
-8.17 |
17 |
|
50 |
1-291 |
54.18 |
-42.44 |
1.07 |
10.69 |
87 |
|
51 |
264-292 |
50.80 |
-44.43 |
2.94 |
3.45 |
47 |
|
52 |
264-276 |
61.41 |
-43.10 |
0.81 |
17.52 |
158 |
|
53 |
264-294 |
61.61 |
-43.11 |
5.40 |
13.12 |
108 |
|
54 |
264-295 |
60.16 |
-44.57 |
7.47 |
8.14 |
70 |
|
55 |
264-296 |
61.77 |
-43.91 |
7.23 |
10.65 |
87 |
|
56 |
264-317 |
64.63 |
-45.10 |
7.17 |
12.38 |
101 |
|
57 |
264-298 |
62.55 |
-46.02 |
9.90 |
6.65 |
62 |
|
58 |
264-299 |
63.18 |
-44.41 |
8.27 |
10.52 |
86 |
|
59 |
264-300 |
57.90 |
-40.23 |
6.24 |
11.45 |
93 |
|
60 |
264-301 |
71.05 |
-40.09 |
8.70 |
22.28 |
238 |
|
61 |
264-302 |
60.84 |
-45.03 |
6.99 |
8.84 |
74 |
|
62 |
264-303 |
60.94 |
-42.51 |
4.81 |
13.64 |
113 |
|
63 |
264-304 |
49.36 |
-42.24 |
3.81 |
3.33 |
46 |
|
64 |
264-305 |
72.03 |
-62.97 |
8.07 |
1.01 |
38 |
|
65 |
264-306 |
56.33 |
-41.32 |
2.81 |
12.22 |
100 |
|
66 |
264-307 |
61.59 |
-40.35 |
1.25 |
20.01 |
196 |
|
67 |
265-308 |
61.92 |
-43.56 |
6.50 |
11.88 |
97 |
|
68 |
266-308 |
63.69 |
-43.81 |
9.64 |
10.26 |
84 |
|
69 |
267-308 |
66.24 |
-44.24 |
10.30 |
11.72 |
96 |
|
70 |
268-308 |
65.69 |
-44.08 |
9.74 |
11.89 |
97 |
|
71 |
269-308 |
63.52 |
-44.01 |
12.39 |
7.14 |
64 |
|
72 |
276-309 |
70.72 |
-59.81 |
12.30 |
-1.37 |
31 |
|
73 |
264-310 |
74.84 |
-47.30 |
2.58 |
24.98 |
301 |
|
74 |
264-311 |
41.66 |
-44.58 |
4.62 |
-7.52 |
18 |
|
75 |
264-312 |
30.29 |
-40.08 |
1.05 |
-10.82 |
14 |
|
76 |
264-313 |
48.91 |
-39.40 |
9.05 |
0.48 |
36 |
|
77 |
264-314 |
75.97 |
-46.86 |
-4.94 |
34.07 |
661 |
|
78 |
264-315 |
78.46 |
-46.91 |
-6.27 |
37.84 |
917 |
|
79 |
264-316 |
75.40 |
-46.35 |
-4.43 |
33.50 |
629 |
|
80 |
264-317 |
76.35 |
-46.80 |
-1.74 |
31.31 |
521 |
|
81 |
264-318 |
77.50 |
-45.72 |
-3.80 |
35.60 |
755 |
|
82 |
264-319 |
73.72 |
-45.80 |
-3.61 |
31.55 |
532 |
|
83 |
264-320 |
80.67 |
-45.84 |
-0.34 |
35.19 |
729 |
|
84 |
264-321 |
76.56 |
-45.65 |
-0.98 |
31.91 |
548 |
|
85 |
264-322 |
64.65 |
-45.79 |
-6.09 |
24.97 |
301 |
|
86 |
264-323 |
65.76 |
-44.75 |
-7.04 |
28.07 |
393 |
|
87 |
264-324 |
65.41 |
-44.98 |
-8.28 |
28.73 |
416 |
|
88 |
264-325 |
64.25 |
-43.28 |
-7.20 |
28.19 |
397 |
|
89 |
264-326 |
65.72 |
-45.70 |
-3.26 |
23.30 |
260 |
|
90 |
264-327 |
64.40 |
-44.77 |
-4.18 |
23.83 |
272 |
|
91 |
264-328 |
64.96 |
-44.53 |
-3.69 |
24.14 |
280 |
|
92 |
270-329 |
78.28 |
-44.77 |
13.70 |
19.83 |
193 |
|
93 |
1-330 |
50.29 |
-44.73 |
8.77 |
-3.19 |
26 |
|
94 |
1-331 |
49.02 |
-43.63 |
9.35 |
-3.94 |
25 |
|
95 |
1-332 |
49.22 |
-43.71 |
5.66 |
-0.13 |
34 |
|
96 |
1-333 |
49.01 |
-44.27 |
5.20 |
-0.44 |
33 |
|
97 |
1-334 |
47.43 |
-39.78 |
1.74 |
5.93 |
58 |
|
98 |
1-335 |
48.25 |
-39.85 |
4.28 |
4.14 |
50 |
|
99 |
1-336 |
61.44 |
-43.86 |
9.71 |
7.89 |
69 |
|
100 |
1-337 |
52.42 |
-44.78 |
8.84 |
-1.18 |
31 |
|
101 |
1-338 |
53.78 |
-42.08 |
8.67 |
3.05 |
45 |
|
102 |
1-339 |
49.79 |
-42.19 |
10.11 |
-2.49 |
28 |
|
103 |
1-340 |
52.37 |
-43.30 |
12.86 |
-3.77 |
25 |
|
104 |
1-341 |
54.50 |
-43.83 |
17.90 |
-7.21 |
19 |
|
105 |
1-342 |
52.62 |
-43.16 |
14.66 |
-5.18 |
22 |
|
106 |
1-343 |
50.79 |
-43.73 |
14.17 |
-7.09 |
19 |
|
107 |
1-344 |
46.63 |
-41.23 |
12.18 |
-6.76 |
19 |
|
108 |
1-345 |
49.63 |
-41.52 |
13.56 |
-5.43 |
22 |
|
109 |
1-346 |
53.91 |
-43.27 |
16.79 |
-6.13 |
20 |
|
110 |
1-347 |
53.47 |
-42.96 |
15.71 |
-5.18 |
22 |
|
111 |
1-348 |
51.92 |
-43.81 |
15.06 |
-6.93 |
19 |
|
112 |
1-349 |
55.41 |
-44.26 |
17.39 |
-6.22 |
20 |
|
113 |
1-350 |
53.23 |
-43.19 |
16.28 |
-6.22 |
20 |
|
114 |
1-351 |
49.40 |
-43.20 |
14.52 |
-8.30 |
17 |
|
115 |
1-352 |
60.46 |
-43.25 |
16.30 |
0.93 |
38 |
|
116 |
1-353 |
58.34 |
-42.83 |
16.34 |
-0.81 |
32 |
|
117 |
1-354 |
60.89 |
-43.36 |
15.88 |
1.67 |
40 |
|
118 |
1-355 |
58.10 |
-42.44 |
17.14 |
-1.46 |
31 |
|
119 |
1-356 |
53.69 |
-43.95 |
14.48 |
-4.72 |
23 |
|
120 |
1-357 |
61.74 |
-42.46 |
14.69 |
4.61 |
52 |
|
121 |
1-358 |
55.78 |
-43.77 |
13.90 |
-1.87 |
29 |
|
122 |
1-359 |
51.57 |
-43.30 |
10.86 |
-2.57 |
28 |
|
123 |
1-360 |
51.86 |
-43.77 |
8.05 |
0.06 |
35 |
|
124 |
1-361 |
56.82 |
-40.62 |
10.51 |
5.71 |
57 |
|
125 |
1-362 |
58.29 |
-43.98 |
12.88 |
1.45 |
39 |
|
126 |
1-363 |
54.12 |
-43.41 |
9.90 |
0.83 |
37 |
|
127 |
271-341 |
60.46 |
-43.98 |
19.36 |
-2.86 |
27 |
|
128 |
271-364 |
65.65 |
-42.46 |
18.59 |
4.62 |
52 |
|
129 |
1-365 |
49.56 |
-42.99 |
12.83 |
-6.24 |
20 |
|
130 |
1-366 |
52.28 |
-43.31 |
14.98 |
-5.99 |
21 |
|
131 |
1-367 |
58.49 |
-44.22 |
13.31 |
0.98 |
38 |
|
132 |
1-368 |
58.63 |
-42.98 |
14.80 |
0.87 |
37 |
|
133 |
1-369 |
49.63 |
-43.63 |
12.72 |
-6.70 |
19 |
|
134 |
1-370 |
50.83 |
-43.67 |
14.48 |
-7.30 |
18 |
|
135 |
1-371 |
52.32 |
-42.65 |
13.67 |
-3.98 |
25 |
|
136 |
1-372 |
58.49 |
-42.17 |
18.74 |
-2.40 |
28 |
|
137 |
1-373 |
55.02 |
-44.44 |
12.21 |
-1.61 |
30 |
|
138 |
1-374 |
54.18 |
-43.72 |
6.74 |
3.74 |
48 |
|
139 |
1-375 |
60.34 |
-43.18 |
12.83 |
4.35 |
50 |
|
140 |
1-376 |
65.18 |
-41.20 |
8.97 |
15.03 |
127 |
|
141 |
1-377 |
65.34 |
-40.83 |
7.78 |
16.75 |
148 |
|
142 |
1-378 |
61.41 |
-40.88 |
6.95 |
13.60 |
112 |
|
143 |
1-379 |
66.62 |
-42.09 |
7.56 |
16.99 |
151 |
|
144 |
1-380 |
61.50 |
-40.61 |
6.66 |
14.25 |
119 |
|
145 |
1-381 |
67.90 |
-37.44 |
9.77 |
20.71 |
208 |
|
146 |
272-382 |
54.53 |
-47.13 |
13.05 |
-5.63 |
21 |
|
147 |
272-383 |
55.40 |
-46.10 |
6.44 |
2.88 |
44 |
|
148 |
272-384 |
59.79 |
-48.03 |
11.71 |
0.07 |
35 |
|
149 |
272-385 |
48.33 |
-47.57 |
7.82 |
-7.04 |
19 |
|
150 |
273-386 |
46.77 |
-42.26 |
7.91 |
-3.38 |
26 |
|
151 |
273-387 |
44.60 |
-43.53 |
12.18 |
-11.09 |
13 |
|
152 |
273-388 |
46.31 |
-45.30 |
13.25 |
-12.22 |
12 |
|
153 |
273-389 |
53.22 |
-45.51 |
5.65 |
2.08 |
41 |
|
154 |
273-390 |
62.97 |
-44.27 |
5.51 |
13.21 |
109 |
|
155 |
273-391 |
51.65 |
-44.95 |
10.96 |
-4.24 |
24 |
|
156 |
273-392 |
50.15 |
-43.65 |
11.12 |
-4.60 |
23 |
|
157 |
273-393 |
46.95 |
-44.81 |
17.60 |
-15.44 |
9 |
|
158 |
273-394 |
43.84 |
-41.72 |
10.04 |
-7.90 |
17 |
|
159 |
273-395 |
52.58 |
-43.29 |
11.57 |
-2.26 |
28 |
|
160 |
273-396 |
46.87 |
-43.93 |
11.36 |
-8.40 |
17 |
|
161 |
273-398 |
52.76 |
-44.82 |
16.58 |
-8.62 |
16 |
|
162 |
273-399 |
48.56 |
-45.20 |
15.58 |
-12.20 |
12 |
|
163 |
273-397 |
46.01 |
-44.64 |
16.22 |
-14.83 |
10 |
|
164 |
273-400 |
46.16 |
-43.80 |
16.19 |
-13.81 |
10 |
|
165 |
273-401 |
59.26 |
-46.14 |
12.43 |
0.71 |
37 |
|
166 |
273-402 |
53.71 |
-47.41 |
14.96 |
-8.64 |
16 |
|
167 |
273-403 |
45.04 |
-43.85 |
9.42 |
-8.21 |
17 |
|
168 |
273-404 |
45.03 |
-45.36 |
16.30 |
-16.61 |
8 |
|
169 |
273-405 |
47.20 |
-42.53 |
13.46 |
-8.77 |
16 |
|
170 |
273-406 |
45.49 |
-45.50 |
13.16 |
-13.15 |
11 |
|
171 |
273-407 |
52.77 |
-46.34 |
17.59 |
-11.14 |
13 |
|
172 |
273-408 |
61.55 |
-49.54 |
8.92 |
3.11 |
45 |
|
173 |
273-409 |
60.82 |
-49.43 |
9.14 |
2.27 |
42 |
|
174 |
273-410 |
50.12 |
-45.44 |
19.36 |
-14.66 |
10 |
|
175 |
274-404 |
44.09 |
-45.26 |
17.25 |
-18.40 |
7 |
|
176 |
275-404 |
-32.78 |
3.04 |
21.04 |
-50.78 |
0.43 |
|
177 |
273-411 |
45.87 |
-46.59 |
17.03 |
-17.73 |
7 |
|
178 |
273-412 |
58.49 |
-51.84 |
11.31 |
-4.64 |
23 |
|
179 |
273-413 |
66.91 |
-44.17 |
17.59 |
5.17 |
54 |
|
180 |
273-414 |
56.95 |
-43.08 |
7.63 |
6.26 |
60 |
|
181 |
273-415 |
56.95 |
-44.82 |
5.69 |
6.46 |
61 |
|
182 |
273-413 |
57.45 |
-44.73 |
3.54 |
9.20 |
77 |
|
183 |
273-417 |
61.86 |
-41.09 |
7.99 |
12.80 |
105 |
|
184 |
275-418 |
55.52 |
-40.35 |
12.49 |
2.70 |
44 |
|
185 |
275-419 |
-19.68 |
5.51 |
20.73 |
-34.89 |
2 |
|
186 |
275-411 |
44.55 |
-46.02 |
16.29 |
-17.74 |
7 |
|
187 |
275-420 |
62.75 |
-44.91 |
6.03 |
11.83 |
96 |
|
188 |
275-421 |
66.35 |
-46.14 |
10.32 |
9.91 |
82 |
|
189 |
275-422 |
67.73 |
-45.16 |
11.45 |
11.14 |
91 |
|
190 |
275-423 |
65.59 |
-45.76 |
8.21 |
11.64 |
95 |
|
191 |
275-424 |
60.50 |
-44.87 |
1.27 |
14.38 |
120 |
|
192 |
275-425 |
59.93 |
-45.31 |
8.10 |
6.54 |
61 |
|
193 |
275-426 |
64.19 |
-45.61 |
5.04 |
13.56 |
112 |
|
194 |
275-427 |
63.09 |
-43.88 |
8.18 |
11.05 |
90 |
|
195 |
275-428 |
64.18 |
-45.35 |
7.73 |
11.12 |
91 |
|
196 |
275-429 |
63.07 |
-46.54 |
8.58 |
7.97 |
69 |
|
197 |
275-430 |
62.86 |
-46.78 |
10.77 |
5.33 |
55 |
|
198 |
275-431 |
52.58 |
-46.71 |
25.47 |
-19.58 |
6 |
|
199 |
275-432 |
47.38 |
-45.03 |
17.59 |
-15.22 |
9 |
|
200 |
275-433 |
55.65 |
-43.97 |
4.07 |
7.63 |
67 |
|
201 |
275-434 |
66.09 |
-42.13 |
8.94 |
15.04 |
127 |
|
202 |
275-435 |
68.27 |
-41.84 |
11.03 |
15.42 |
132 |
|
203 |
275-436 |
57.69 |
-42.12 |
11.22 |
4.37 |
51 |
|
204 |
275-437 |
57.12 |
-39.57 |
12.87 |
4.70 |
52 |
|
205 |
275-438 |
56.79 |
-41.44 |
13.60 |
1.77 |
40 |
|
206 |
275-439 |
51.88 |
-48.48 |
19.73 |
-16.31 |
8 |
|
207 |
275-440 |
-21.06 |
5.19 |
16.43 |
-32.30 |
2 |
|
208 |
275-441 |
47.50 |
-45.02 |
13.79 |
-11.29 |
13 |
|
209 |
275-442 |
56.00 |
-34.52 |
15.54 |
5.96 |
58 |
|
210 |
275-443 |
64.12 |
-36.88 |
17.10 |
10.16 |
83 |
|
211 |
22-444 |
66.60 |
-46.64 |
-0.66 |
20.64 |
207 |
|
212 |
22-420 |
65.46 |
-47.14 |
-0.77 |
19.11 |
181 |
|
213 |
22-445 |
66.82 |
-45.87 |
4.23 |
16.74 |
147 |
|
214 |
22-446 |
65.85 |
-47.85 |
2.09 |
15.93 |
138 |
|
215 |
22-447 |
63.91 |
-46.13 |
1.90 |
15.90 |
137 |
|
216 |
22-448 |
65.29 |
-45.88 |
-1.47 |
20.90 |
211 |
|
217 |
275-449 |
-28.13 |
-0.71 |
20.88 |
-49.71 |
0.47 |
|
218 |
275-450 |
52.58 |
-48.90 |
11.95 |
-8.25 |
17 |
|
219 |
275-451 |
59.14 |
-50.53 |
15.01 |
-6.38 |
20 |
|
220 |
275-452 |
-24.41 |
-2.25 |
22.80 |
-49.45 |
0.48 |
|
221 |
275-453 |
-15.22 |
0.01 |
20.57 |
-35.79 |
2 |
|
222 |
22-454 |
68.00 |
-44.22 |
4.80 |
19.00 |
179 |
|
223 |
22-455 |
67.92 |
-44.47 |
3.33 |
20.14 |
198 |
|
224 |
275-456 |
45.00 |
-43.59 |
12.85 |
-11.42 |
13 |
|
225 |
275-457 |
56.31 |
-48.43 |
10.66 |
-2.76 |
27 |
|
226 |
275-458 |
60.02 |
-47.92 |
25.25 |
-13.13 |
11 |
|
227 |
275-459 |
58.25 |
-47.42 |
18.25 |
-7.40 |
18 |
|
228 |
275-460 |
43.69 |
-47.26 |
21.73 |
-25.28 |
4 |
|
229 |
275-461 |
44.72 |
-45.89 |
19.94 |
-21.09 |
6 |
aDDHMM is the relative enthalpic contribution to the GFE change of the HDAC8-DAHT complex formation DDGcom (for details see footnote of Table 2);
bDDGsol is the relative solvation GFE contribution to DDGcom; cDDTSvib is the relative (vibrational) entropic contribution to DDGcom;
dDDGcom is the relative Gibbs free energy change related to the enzyme- inhibitor HDAC8-DAHT complex formation DDGcom @ DDHMM + DDGsol - DDTSvib.
e IC50pre is the predicted inhibition potency towards HDAC8 calculated from DDGcom using correlation equation A, Table 3;
f IC50exp 25 is given for the reference inhibitor DAHT1 instead of the .
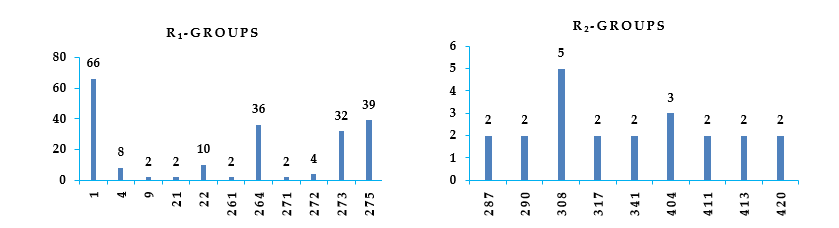
Figure 6. Histograms of frequency of occurrence of individual R-groups in the 229 best selected analogues mapping to features of the PH4 pharmacophore hypothesis Hypo1 (for the structures of the fragments see Table 5).
3.6. Substituent impact on binding mode of novel DAHT analogues
The design of virtual library of novel analogues was guided by structural information retrieved from the DAHTx active conformation and was used for the selection of appropriate substituents (R1-R2-groups). In order to identify which substituents, lead to new inhibitor candidates with the highest predicted potencies towards the HDAC8, we have prepared histograms of the frequency of occurrence of R1-R2-groups among the 229 best fit PH4 hits (Figure 6). Analysis of the histograms showed that the highest frequency of occurrence among the R1-groups displayed the fragments 1(66), 4(8), 22(10), 264(36), 273(32) and 275(39). In case of R2 groups 308(5) and 404(3).
An analysis of structural requirement for human HDAC8 inhibition at the level of hydrophobic contacts with the active site revealed that the P2 substituent, namely the R2-group in the training set insufficiently explored the S2 sub-pocket of the active site. Therefore, new DAHT analogues that match the HDAC8 inhibition pharmacophore and fill better the S2 sub-pocket may form potent HDAC8 inhibitors (Table 6). The top scoring virtual hits are DAHT analogues: 274-404 (IC50pre = 0.43 nM), 275-449 (IC50pre = 0.47 nM), 273-452 (IC50pre = 0.48 nM). The best analogue designed 275-404 ( = 0.43 nM) displays predicted potency approximately 110 times better than the best of training set compound DAHT1 (IC50pre = 47 nM). Our approach helped to identify interesting hydrophobic side chains (R1-groups) such as indol-2H-yl (273), 6-methoxy-1H-indol-2-yl (274) and 5,6-dimethoxy-1H-indol-2-yl (275) for the filling of the S1 sub-pocket with a bulkier group compared to the training set inhibitors, which contain for most of the inhibitors only the 4-methoxyphenyl group in the P1 position.
Figure 7.c, d show π-π stacking interactions between the hydrophobic group 5,6-dimethoxy-1H-indol-2-yl and the residue Tyr100, which are stabilizing in nature [60]. As we can see on Figure 7, the three best analogue designs are shown.
Our approach also allowed us to identify side chains (R2-groups), which are the most bulky but most specific to S2 sub-pocket such as 4-(((2R,3R,4S,6R)-2-(tert-butyl)-4-(1-chloro-3-fluoro-2-methylpropan-2-yl)-3-ethoxy-6-fluorocyclohexyl)amino)-3-(6,6-dimethylpiperidin-1-ium-2-carboxamido)-2-(4-(2-fluoroethyl)phenyl)pentanoyl (452); 3-((3-ammonio-3-methylcyclohexyl)carbamoyl)-4-(((2R,3R,4S,6R)-2-(tert-butyl)-4-(1-chloro-3-fluoro-2-methylpropan-2-yl)-3-ethoxy-6-fluorocyclohexyl)amino)-2-(4-(2-fluoroethyl)phenyl)pentanoyl (404); (4-(((2R,3R,4S,6R)-2-(tert-butyl)-4-(1-chloro-3-fluoro-2-methylpropan-2-yl)-3-ethoxy-6-fluorocyclohexyl)amino)-3-((6,6-dimethylpiperidin-1-ium-2-yl)carbamoyl)-2-(4-(2-fluoroethyl)phenyl)pentanoyl (449), Table 5. Indeed, Figure 8 shows the increase in the affinity, through interaction energy between catalytic residues (Tyr100, Asp101) of S1 sub-pocket and the hydrophobic group 5,6-dimethoxy-1H-indol-2-yl of one the best-designed analogues 275-404 ( IC50pre = 0.43n M) compared to the most active training set inhibitor DAHT1.
According to our analysis of the HDAC8-DAHTx complexes of the most potent inhibitors, several interactions play a key role in the significant improvement of predicted inhibitory potencies of the novel tetrahydroisoquinoline-based hydroxamic acid derivatives. Based on the intermolecular interaction energy breakdown to residue contributions (Figure 4), the residues Phe207 and Pro273; in addition to the catalytic residues Tyr100 and Met274 residues, play an important role in the inhibition of HDAC8. According to Tabackman et al. [26], the Pro273 residue is known to create van der Waals interactions with HDAC8 inhibitors such as SAHA which is consistent with the data from our study (Figure 8.B). We have observed a π-π stacking interaction between the phenyl group of the best-designed analogue 275-404 and Phe207 residue (Figure 7.c, d). Substitution of the 2,2-dimethylpiperid-6-yl group (analogue 275-404) in place of the pyrrolid-2-yl group (DAHT1 training set) significantly increases affinity of the analogue to HDAC8. Indeed, the 2,2-dimethylpiperid-6-yl group has a greater affinity with the residues Pro273, Met274 and the cofactor UNK405H compared to the pyrrolid-2-yl group. Thus, we have a π-donor hydrogen bond interaction between the phenyl group of the DAHT scaffold and the catalytic residue Phe208. The fragment (404) R2-group of the best analogue designed allows the scaffold to adopt a favourable position with respect to the catalytic residue Phe208 thus increasing the affinity with HDAC8 (Figure 8.B). Other residues of the HDAC8 active site such as Lys202 and Arg356 also contribute to high activity of novel analogues designed (see Figure 8.).
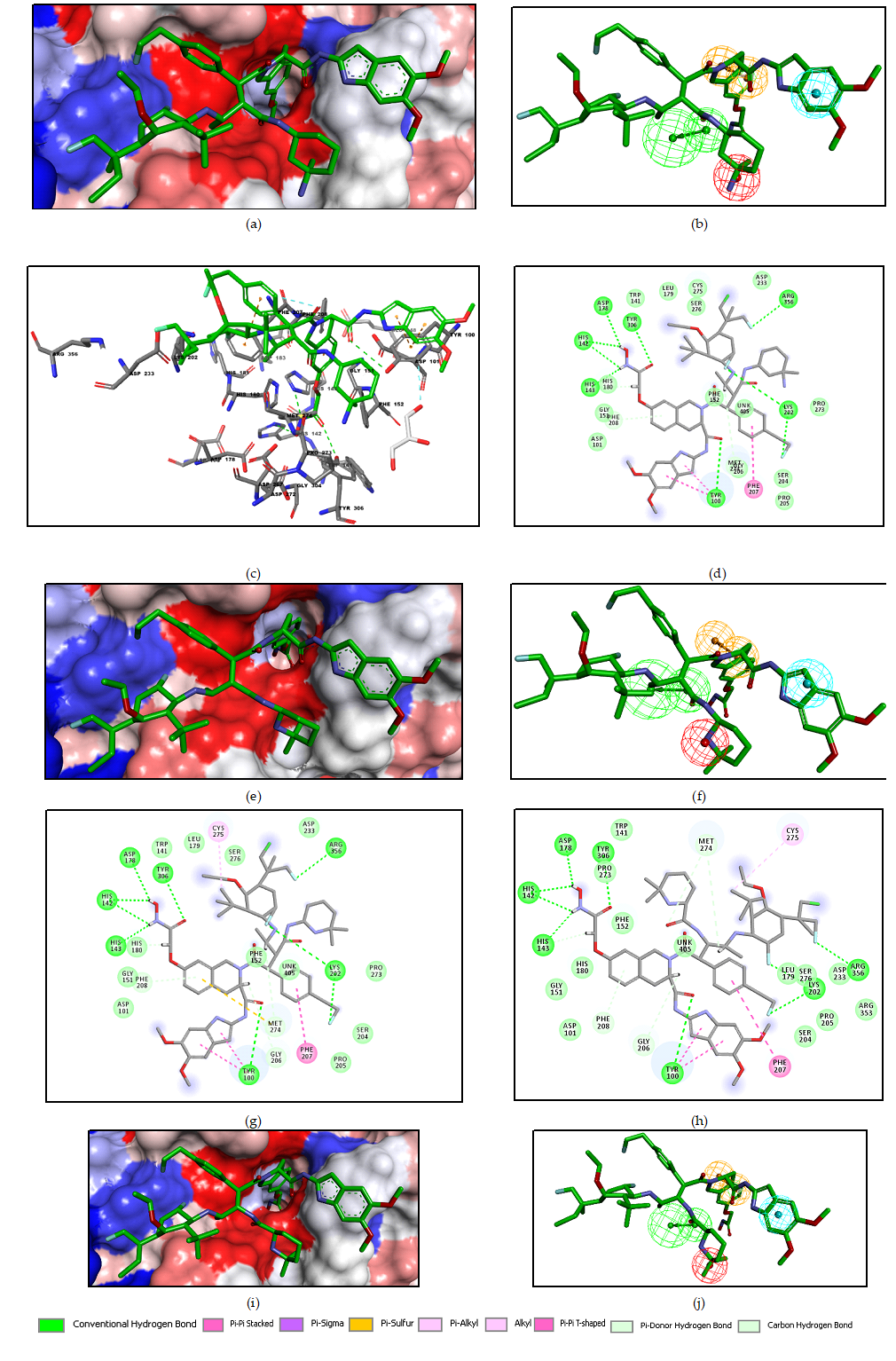
Figure 7. (a) - Connolly surface of the active site of HDAC8 with bound most active designed DAHT analogue 275-404 ( = 0.43 nM). The binding site surface is coloured according to residue hydrophobicity: red - hydrophobic, blue - hydrophilic and white - intermediate. (b) - mapping of the DAHT 275-404 to HDAC8 inhibition pharmacophore. (c) - close up of virtual hit DAHT 275-404 at the active site of HDAC8. (d) - 2D schematic interaction diagram of the DAHT 275-404 at the active site of HDAC8. (e) - Connolly surface of the active site of HDAC8 with bound DAHT analogue 275-449 (IC50pre = 0.47 nM). (f) - mapping of the DAHT 275-449 to HDAC8 inhibition pharmacophore. (g) - 2D schematic interaction diagram of the analogue DAHT 275-449 ( IC50pre= 0.47 nM) at the active site of HDAC8. (h)- 2D schematic interaction diagram of the analogue DAHT 275-452 ( IC50pre = 0.48 nM) at the active site of HDAC8. (i) - Connolly surface of the active site of HDAC8 with bound DAHT analogue 275-452 (IC50pre = 0.48 nM). (j) - mapping of the DAHT 275-452 to HDAC8 inhibition pharmacophore.
3.7. Pharmacokinetic profile of novel DAHT analogues
Among the ADME-related properties displayed in Table 7, such as octanol-water partitioning coefficient, aqueous solubility, blood-brain partition coefficient, Caco-2 cell permeability, serum protein binding, number of likely metabolic reactions, and another eighteen descriptors related to absorption, distribution, metabolism and excretion (ADME) of the new analogues were computed by the QikProp program [55] based on the method of Jorgensen [56,57]. Experimental data from more than 710 compounds including about 500 drugs and related heterocycles were used to produce regression equations correlating experimental and computed descriptors resulting in an accurate prediction of pharmacokinetic properties of molecules. Drug likeness (#stars) - the number of property descriptors that fall outside the range of optimal values determined for 95% of known drugs out of 24 selected descriptors computed by the QikProp, was used as an additional ADME-related compound selection criterion. The values for the best active designed DAHTs are compared with those computed for drugs used for treatment of cancer or currently undergoing clinical trials, Table 7. It can be noted that human oral absorption through the gastrointestinal system (HOA) is low for our best designed analogues suggesting non-oral delivery. The descriptor of the blood-brain barrier is within the appropriate range.
Table 7. Predicted ADME-related properties of the best designed DAHT analogues and known anticancer agents either in clinical use or currently undergoing clinical testing computed by QikProp [55].
|
DAHTx a |
#starsb |
Mwc [g.mol-1] |
Smold [Å2] |
Smol,hfoe [Å2] |
Vmolf [Å3] |
RotBg |
HBdonh |
HBacci |
logPo/wj |
logSwatk |
logKHSAl |
logB/Bm |
BIPcacon [nm.s-1] |
#metao |
IC50pr [nM] |
HOAq |
%HOAr |
|
275-404 |
14 |
1124.8 |
1424.5 |
817.5 |
3091.5 |
22 |
6 |
17 |
7.1 |
-7.5 |
1.373 |
-2.6 |
1.6 |
11 |
0.43 |
1 |
20.4 |
|
275-449 |
15 |
1124.8 |
1423.3 |
864.7 |
3109.3 |
21 |
4 |
16 |
8.3 |
-8.2 |
1.816 |
-2.0 |
3.6 |
11 |
0.47 |
1 |
33.3 |
|
275-452 |
15 |
1124.8 |
1426.0 |
858.1 |
3112.5 |
21 |
5 |
17 |
7.7 |
-7.6 |
1.551 |
-2.0 |
3.4 |
11 |
0.48 |
1 |
29.6 |
|
275-453 |
14 |
1124.8 |
1416.8 |
814.6 |
3094.6 |
22 |
6 |
17 |
7.1 |
-7.2 |
1.388 |
-2.5 |
1.6 |
11 |
2 |
1 |
20.4 |
|
275-419 |
15 |
1152.8 |
1434.1 |
864.0 |
3171.6 |
23 |
6 |
17 |
7.8 |
-7.5 |
1.554 |
-2.1 |
3.3 |
12 |
2 |
1 |
30.3 |
|
275-440 |
13 |
1139.8 |
1436.7 |
884.6 |
3121.9 |
22 |
6 |
17 |
6.7 |
-6.4 |
1.45 |
-2.5 |
0.5 |
12 |
2 |
1 |
9.0 |
|
1-351 |
8 |
921.5 |
1099.3 |
625.5 |
2484.9 |
18 |
5 |
15 |
4.8 |
-5. 1 |
0.096 |
-2.4 |
23.4 |
9 |
17 |
1 |
52.3 |
|
273-394 |
9 |
1094.7 |
1284.4 |
586.9 |
2908.1 |
20 |
6 |
16 |
6.2 |
-6.1 |
0.877 |
-2.7 |
2.4 |
10 |
17 |
1 |
18.1 |
|
264-290 |
9 |
795.3 |
892.1 |
518.9 |
2025.9 |
23 |
9 |
19 |
-1.1 |
2.0 |
-1.389 |
-1.6 |
1.1 |
15 |
17 |
1 |
0 |
|
1-343 |
8 |
825.1 |
1021.3 |
644.1 |
2316.3 |
16 |
5 |
14 |
4.1 |
-4.6 |
0.088 |
-2.2 |
38.6 |
9 |
19 |
1 |
53.2 |
|
1-344 |
8 |
843.1 |
1037.2 |
644.7 |
2320.2 |
16 |
5 |
14 |
4.5 |
-5.4 |
0.080 |
-1.7 |
104.4 |
9 |
19 |
1 |
63.6 |
|
1-341 |
9 |
851.1 |
1095.8 |
789.6 |
2467.1 |
18 |
5 |
15 |
4.4 |
-4.6 |
0.048 |
-2.1 |
56.9 |
9 |
19 |
1 |
58.3 |
|
1-346 |
8 |
869.1 |
1121.4 |
690.4 |
2442.9 |
18 |
5 |
15 |
4.1 |
-5.3 |
-0.053 |
-2.6 |
27.4 |
9 |
20 |
1 |
50.8 |
|
1-349 |
8 |
869.1 |
1031.6 |
698.8 |
2377.3 |
18 |
5 |
15 |
4.1 |
-4.6 |
-0.127 |
-1.8 |
147.1 |
9 |
20 |
1 |
63.9 |
|
1-350 |
8 |
887.1 |
1028.9 |
650.6 |
2404.7 |
18 |
5 |
15 |
4.3 |
-3.9 |
-0.044 |
-1.8 |
79.7 |
9 |
20 |
1 |
60.4 |
|
1-365 |
8 |
936.6 |
1153.7 |
671.9 |
2537.9 |
19 |
6 |
15 |
3.9 |
-3.5 |
0.082 |
-2.5 |
1.7 |
9 |
20 |
1 |
15.1 |
|
SAHA |
0 |
264.3 |
560 |
204.0 |
939 |
9 |
3 |
7 |
0.7 |
-1.3 |
-0.807 |
-1.5 |
134.8 |
3 |
1480 |
2 |
69.0 |
|
Valproic acid |
3 |
144.2 |
392 |
311.0 |
621 |
5 |
1 |
2 |
2.7 |
-1.9 |
-0.45 |
-0.4 |
431.8 |
1 |
|
3 |
90.2 |
|
Givinostat |
0 |
407.5 |
768 |
263.0 |
1330 |
8 |
3 |
8 |
3.1 |
-6.0 |
0.277 |
-2.2 |
140.6 |
2 |
|
3 |
83.6 |
|
Sodium phenylbutyrate |
0 |
164.2 |
402 |
99.9 |
636 |
4 |
1 |
2 |
2.1 |
-1.8 |
-0.371 |
-0.6 |
238.6 |
2 |
|
3 |
81.7 |
|
R306465 |
1 |
413.5 |
686 |
143.3 |
1194 |
4 |
2 |
11 |
1.3 |
-4.3 |
-0.449 |
-1.6 |
190.7 |
2 |
|
3 |
75.1 |
|
Cra024781 |
0 |
397.4 |
715 |
222.8 |
1249 |
9 |
3 |
10 |
1.4 |
-3.1 |
-0.308 |
-1.5 |
49.3 |
4 |
|
3 |
65.3 |
|
Entinostat |
2 |
376.4 |
736 |
70.2 |
1239 |
7 |
4 |
8 |
2.9 |
-5.6 |
0.091 |
-1.8 |
247.8 |
8 |
|
3 |
87.0 |
|
Mocetinostat |
3 |
396.5 |
729 |
36.0 |
1263 |
7 |
4 |
8 |
3.3 |
-5.5 |
0.169 |
-1.5 |
422.1 |
9* |
|
3 |
93.4 |
|
Pivanex |
1 |
202.3 |
482 |
408.3 |
791 |
5 |
0 |
4 |
2.0 |
-2.4 |
-0.368 |
-0.4 |
1986.4 |
1 |
|
3 |
100.0 |
|
Pracinostat |
0 |
358.5 |
732 |
444.5 |
1279 |
12 |
2 |
8 |
2.5 |
-3.6 |
0.037 |
-1.4 |
90.5 |
2 |
|
2 |
76.8 |
|
Tacedinaline |
0 |
269.3 |
528 |
74.9 |
890 |
4 |
4 |
6 |
1.2 |
-3.1 |
-0.298 |
-1.3 |
225.6 |
3 |
|
3 |
76.0 |
|
Romidepsin |
15 |
787.6* |
1055* |
307.6 |
1981 |
20* |
10* |
27* |
-2.9* |
-3.0 |
-2.32* |
-7.9* |
0.0 |
11* |
|
1 |
0.0 |
|
Belinostat |
0 |
318.3 |
568 |
26.5 |
979 |
8 |
3 |
9 |
0.7 |
-1.2 |
-0.84 |
-2.2 |
0.5 |
1 |
|
1 |
26.3 |
|
Panobinostat |
1 |
349.4 |
636 |
210.1 |
1150 |
9 |
3 |
7 |
1.7 |
-2.6 |
-0.055 |
-1.4 |
43.3 |
6 |
|
2 |
66.4 |
a designed DAHT analogues and known anticancer agents, Tables 6;
b drug likeness, number of property descriptors (24 out of the full list of 49 descriptors of QikProp, ver. 3.7, release 14) that fall outside of the range of values for 95% of known drugs;
c molecular weight in [g.mol-1] (range for 95% of drugs: 130 - 725 g.mol-1) [61];
d total solvent-accessible molecular surface, in [Å2] (probe radius 1.4 Å) (range for 95% of drugs: 300 - 1000 Å2);
e hydrophobic portion of the solvent-accessible molecular surface, in [Å2] (probe radius 1.4 Å) (range for 95% of drugs: 0 - 750 Å2);
f total volume of molecule enclosed by solvent-accessible molecular surface, in [Å3] (probe radius 1.4 Å) (range for 95% of drugs: 500 - 2000 Å3);
g number of non-trivial (not CX3), non-hindered (not alkene, amide, small ring) rotatable bonds (range for 95% of drugs: 0 - 15);
h estimated number of hydrogen bonds that would be donated by the solute to water molecules in an aqueous solution. Values are averages taken over several configurations, so they can assume non-integer values (range for 95% of drugs: 0.0 - 6.0);
i estimated number of hydrogen bonds that would be accepted by the solute from water molecules in an aqueous solution. Values are averages taken over several configurations, so they can assume non-integer values (range for 95% of drugs: 2.0 - 20.0);
j logarithm of partitioning coefficient between n-octanol and water phases (range for 95% of drugs: -2 - 6.5);
k logarithm of predicted aqueous solubility, logS. S in [mol·dm–3] is the concentration of the solute in a saturated solution that is in equilibrium with the crystalline solid (range for 95% of drugs: -6.0 - 0.5);
l logarithm of predicted binding constant to human serum albumin (range for 95% of drugs: -1.5 - 1.5);
m logarithm of predicted brain/blood partition coefficient (range for 95% of drugs: -3.0 - 1.2);
n predicted apparent Caco-2 cell membrane permeability in Boehringer-Ingelheim scale in [nm s-1] (range for 95% of drugs: < 25 poor, > 500 nm s-1 great);
o number of likely metabolic reactions (range for 95% of drugs: 1 - 8);
p predicted inhibition constants IC50pre. The IC50pre was predicted from computed DDGcom using the regression equation B shown in Table 3;
q human oral absorption (1 - low, 2 - medium, 3 - high);
r percentage of human oral absorption in gastrointestinal tract (<25% - poor, >80% high);
(*) star indicating that the property descriptor value falls outside the range of values for 95% of known drugs.
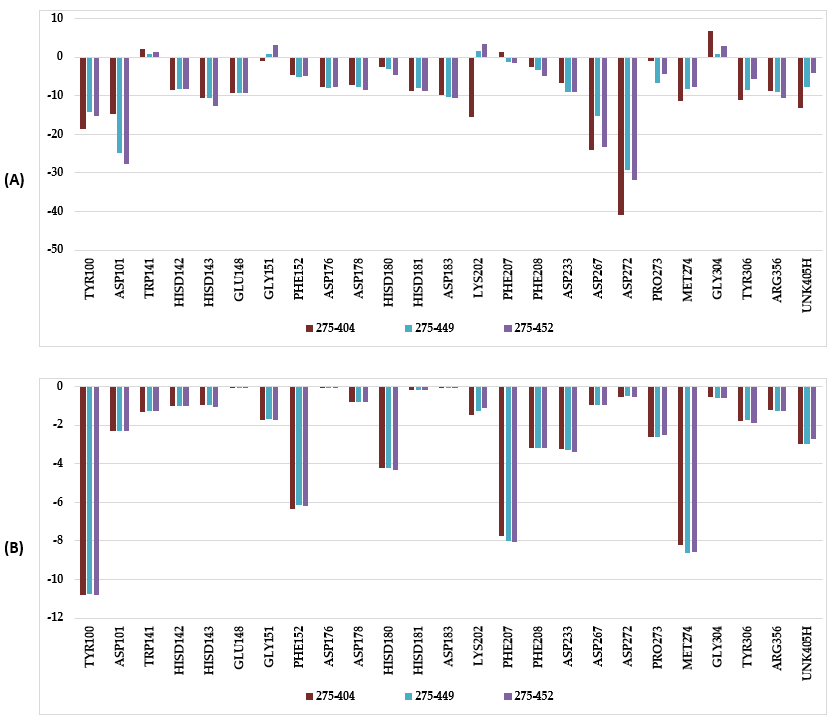
Figure 8. (A) Molecular mechanics intermolecular interaction energy Eint breakdown to residue contributions, in [kcal.mol-1] shown for the best three designed novel DAHT analogues. (B) van-der-Waals component (Eint-vdW) of the molecular mechanics intermolecular interaction energy breakdown to residue contributions in [kcal.mol-1], shown for the best three designed novel DAHT analogues (the color coding refers to ligands and is given in the legend).
Design of new potent DAHT analogues inhibiting human HDAC8 with favourable pharmacokinetic profiles is needed to extend the portfolio of currently available anticancer drugs. Structural information from the crystal structure of the HDAC8-SAHA complex [32] guided us during the development of a reliable QSAR model for the non-covalent inhibition of HDAC8 by tetrahydroisoquinoline-based hydroxamic acid derivatives (DAHT), which correlated the computed Gibbs free energies of complex formation with the observed HDAC8 inhibitory potencies [25]. In addition to this QSAR model, we have elaborated a 3D QSAR pharmacophore model for DAHT inhibitors. Analysis of interactions between HDAC8 and DAHT in the active site of the enzyme was helpful in our effort to design a virtual combinatorial library of new DAHT analogues with multiple substitutions. The design strategy was based mainly on the presence of the hydrophobic features included in the best PH4 pharmacophore models at the P1 and P2 positions of DAHTs. The initial virtual library was screened by matching PH4 pharmacophore analogues and allowed the selection of a focused library subset. The best virtual compounds were subjected to prediction of inhibitory potencies from computed GFE by means of the QSAR model derived from training set of known DAHTs. The best-designed analogues display predicted low nanomolar inhibitory concentrations 274-404 (0.43 nM), 275-449 (0.47 nM), 275-452 (0.48 nM), 275-419 (2 nM), 275-440 (2 nM), 275-453 (2 nM), 275-460 (4 nM) (Table 6). The predicted inhibitory potencies of the best-designed analogues are up to 110 times higher than that of the most active training set inhibitor DAHT1. They are recommended for synthesis and biological evaluation to specialized laboratories in order to develop new anticancer drugs with a promising pharmacokinetic profile.
Partial financial support by the Slovak Research and Development Agency grants (APVV-15-0111, APVV-17-0239 and APVV-18-0420) as well as Granting Agency of Slovak Ministry of Education and Slovak Academy of Sciences (VEGA 1/0228/17) is gratefully acknowledged.
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. 2018 CA CANCER J CLIN. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. 68, 394-424. PMid:30207593
View Article PubMed/NCBIWorld Health Organization. Global Health Observatory, «Geneva: World Health Organization» 21 June 2018. [En ligne]. Available: en/
View ArticleD. Mottet et V. Castronovo. 2008 Les histones désacétylases : nouvelles cibles thérapeutiques anti-cancéreuses. MEDECINE/SCIENCES 24, 742-6. (doi:10.1051/medsci/20082489742) PMid:18789222
View Article PubMed/NCBIN. Archin, K. Keedy, H. Dang, A. Espeseth, D. Hazuda and D. Margolis. 2009 Expression of latent human immunodeficiency type 1 is induced by novel and selective histone deacetylase inhibitors. Aids 23, 1799-1806. (doi:10.1097/QAD.0b013e32832ec1dc) PMid:19590405
View Article PubMed/NCBIK. J. Falkenberg et R. W. Johnstone. 2014 Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 13, 673-691. (doi:10.1038/nrd4360) PMid:25131830
View Article PubMed/NCBIK. Halkidou, L. Gaughan, S. Cook, H. Y. Leung, D. E. Neal and C. N. Robson. 2004 Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate 59, 177-189. (doi:10.1002/pros.20022) PMid:15042618
View Article PubMed/NCBIJ. H. Choi, H. J. Kwon, B. I. Yoon, J. H. Kim, S. U. Han, H. J. Joo and D. Y. Kim. 2001 Expression profile of histone deacetylase 1 in gastric cancer tissues. Jpn. J. Cancer Res. 92, 1300-1304. (doi:10.1111/j.1349-7006.2001.tb02153.x) PMid:11749695
View Article PubMed/NCBIA. J. Wilson, D. S. Byun, N. Popova, L. B. Murray, K. L'Italien, Y. Sowa, D. Arango, A. Velcich, L. H. Augenlicht and J. M. Mariadason. 2006 Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J. Biol. Chem. 281, 13548-13558. (doi:10.1074/jbc.M510023200) PMid:16533812
View Article PubMed/NCBIZ. Zhang, H. Yamashita, T. Toyama, H. Sugiura, Y. Ando, K. Mita, M. Hamaguchi, Y. Hara, S. Kobayashi and H. Iwase. 2005 Quantitation of HDAC1 mRNA expression in invasive carcinoma of the breast. Breast Cancer Res. Treat. 94, 11-16. (doi:10.1007/s10549-005-6001-1) PMid:16172792
View Article PubMed/NCBIP. Zhu, E. Martin, J. Mengwasser, P. Schlag, K. Janssen and M. Göttlicher. 2004 Induction of HDAC2 expression upon loss of APC in colorectal tumorigenesis. Cancer Cell 5, 455-463. (doi: 10.1016/s1535-6108(04)00114-x) 00114-X
View ArticleB. H. Huang, M. Laban, C. H. Leung, L. Lee, C. K. Lee, S.-T. M, G. C. Raju and S. C. Hooi. 2005 Inhibition of histone deacetylase 2 increases apoptosis and p21Cip1/WAF1 expression, independent of histone deacetylase 1. Cell Death Differ 12, 395-404. (doi:10.1038/sj.cdd.4401567) PMid:15665816
View Article PubMed/NCBIJ. Song, J. H. Noh, J. H. Lee, J. W. Eun, Y. M. Ahn, S. Y. Kim, S. H. Lee, W. S. Park, N. J. Yoo, J. Y. Lee and S. W. Nam. 2005 Increased expression of histone deacetylase 2 is found in human gastric cancer. APMIS 113, 264-268. (doi:10.1111/j.1600-0463.2005.apm_04.x) PMid:15865607
View Article PubMed/NCBIZ. Zhang, H. Yamashita, T. Toyama, H. Sugiura, Y. Omoto, Y. Ando, K. Mita, M. Hamaguchi, S. Hayashi and H. Iwase. 2004 Clin. Cancer Res. 10, 6962-6968. (doi:10.1158/1078-0432.CCR-04-0455) PMid:15501975
View Article PubMed/NCBIH. J. Kee and H. Kook. 2011 Roles and targets of class I and IIa histone deacetylases in cardiac hypertrophy. J Biomed Biotechnol 2011, 928326. (doi:10.1155/2011/928326) PMid:21151616
View Article PubMed/NCBIC. Hassig and S. Schreiber. 1997 Nuclear histone acetylases and deacetylases and transcriptional regulation: HATs off to HDACs. Curr. Opin. Chem. Biol. 1, 300-308. (doi:10.1016/s1367-5931(97)80066-x) 80066-X
View ArticleT. Kouzarides. 1999 Histone acetylases and deacetylases in cell proliferation. Curr. Opin. Genet. Dev. 9, 40-48. (doi:10.1016/s0959-437x(99)80006-9) 80006-9
View ArticleA. P. Wolffe. 1996 Histone deacetylase: a regulator of transcription. Science (Washington, DC, U. S.) 272, 371-372. (doi: 10.1126/science.272.5260.371) PMid:8602525
View Article PubMed/NCBIM. Dokmanovic, C. Clarke and P. Marks. 2007 Histone deacetylase inhibitors: overview and perspectives. Mol. Cancer Res. 5, 981- 989. (doi:10.1158/1541-7786.MCR-07-0324) PMid:17951399
View Article PubMed/NCBIS. Ropero and M. Esteller. 2007 The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol. 1, 19-25. (doi:10.1016/j.molonc.2007.01.001) PMid:19383284
View Article PubMed/NCBIX. J. Yang and E. Seto. 2008 The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol 9, 206-218. (doi:10.1038/nrm2346) PMid:18292778
View Article PubMed/NCBIH. Lehrmann, L. L. Pritchard and A. Harel-Bellan. 2002 Histone acetyltransferases and deacetylases in the control of cell proliferation and differentiation. Adv Cancer Res. 86, 41-65. (doi:10.1016/s0065-230x(02)86002-x) 86002-X
View ArticleP. Marks and R. Breslow. 2007 Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat Biotechnol 25, 84-90. (doi:10.1038/nbt1272) PMid:17211407
View Article PubMed/NCBIS. Emiliani, W. Fischle, C. V. Lint, Y. Al-Abed and E. Verdin. 1998 Characterization of a human RPD3 ortholog, HDAC3. Proc Natl Acad Sci. 95, 2795-2800. (doi:10.1073/pnas.95.6.2795) PMid:9501169
View Article PubMed/NCBIA. de Ruijter, A. van Gennip, H. Caron, S. Kemp and A. van Kuilenburg. 2003 Histone deacetylases (HDACs): Characterisation of the classical HDAC family. Biochem. J. 370, 737-749. (doi:10.1042/BJ20021321) PMid:12429021
View Article PubMed/NCBIY. Zhang, J. Feng, Y. Jia, X. Wang, L. Zhang, C. Liu, H. Fang and W. Xu. 2011 Development of Tetrahydroisoquinoline-Based Hydroxamic Acid Derivatives: Potent Histone Deacetylase Inhibitors with Marked in Vitro and in Vivo Antitumor Activities. J. Med. Chem. 54, 2823-2838. (doi:10.1021/jm101605z) PMid:21476600
View Article PubMed/NCBIA. A. Tabackman, R. Frankson, E. S. Marsan, K. Perry and K. E. Colea. 2016 Structure of 'linkerless' hydroxamic acid inhibitor-HDAC8 complex confirms the formation of an isoform-specific subpocket. Journal of Structural Biology 195, 373-378. (doi:10.1016/j.jsb.2016.06.023) PMid:27374062
View Article PubMed/NCBIM. A. Glozak, N. Sengupta, X. Zhang and E. Seto. 2005 Acetylation and deacetylation of non-histone proteins. Gene 363, 15-23. (doi:10.1016/j.gene.2005.09.010) PMid:16289629
View Article PubMed/NCBIY. Zhang, H. Fang, J. Jiao and W. Xu. 2008 The structure and function of histone deacetylases: the target for anti-cancer therapy. Curr. Med. Chem. 15, 2840-2849. (doi:10.2174/092986708786242796) PMid:18991639
View Article PubMed/NCBIY. Zhang, J. Feng, C. Liu, L. Zhang, J. Jiao, H. Fang, L. Su, X. Zhang, J. Zhang, M. Li, B. Wang and W. Xu. 2010 Design, synthesis and preliminary activity assay of 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid derivatives as novel Histone deacetylases (HDACs) inhibitors. Bioorg. Med. Chem. 18, 1761-1772. (doi:10.1016/j.bmc.2010.01.060) PMid:20171895
View Article PubMed/NCBIY. Zhang, H. Fang, J. Feng, Y. Jia, X. Wang, and W. Xu. 2011 Discovery of a Tetrahydroisoquinoline-Based Hydroxamic Acid Derivative (ZYJ-34c) as Histone Deacetylase Inhibitor with Potent Oral Antitumor Activities. J. Med. Chem. 54, 5532-5539. (doi:10.1021/jm200577a) PMid:21714538
View Article PubMed/NCBIB. Bauvois and D. Dauzonne. 2006 Aminopeptidase-N/CD13 (EC 3.4.11.2) inhibitors: chemistry, biological evaluations, and therapeutic prospects. Med. Res. Rev. 26, 88-130. (doi:10.1002/med.20044) PMid:16216010
View Article PubMed/NCBIC. Decroos, C. Bowman, J. Moser, K. Christianson, M. Deardorff and D. Christianson. 2014 Crystal structure of C153F HDAC8 in complex with SAHA. RCSB The Protein Data Bank, 06 08 2014. [online]. Available: . (doi:10.2210/pdb4QA0/pdb) https://doi.org/10.2210/pdb4qa0/pdb
View ArticleM. Keita, A. Kumar, B. Dali, E. Megnassan, M. I. Siddiqi, V. Frecer and S. Miertuš. 2014 Quantitative structure-activity relationships and design of thymine-like inhibitors of thymidine monophosphate kinase of Mycobacterium tuberculosis with favourable pharmacokinetic profiles. RSC Adv. 4, 55853-66. (doi:10.1039/c4ra06917j)
View ArticleA. F. Kouassi, M. Kone, M. Keita, A. Esmel, E. Megnassan, Y. N'Guessan, V. Frecer and S. Miertuš. 2015 Computer-aided design of orally bioavailable pyrrolidine carboxamide inhibitors of enoyl-acyl carrier protein reductase of Mycobacterium tuberculosis with favorable pharmacokinetic profiles. Int J Mol Sci. 16, 29744-71. (doi:10.3390/ijms161226196) PMid:26703572
View Article PubMed/NCBIK. N. P. G. Allangba, M. Keita, R. K. N'Guessan, E. Megnassan, V. Frecer and S. Miertuš. 2019 Virtual design of novel Plasmodium falciparum cysteine protease falcipain-2 hybrid lactone-chalcone and isatin-chalcone inhibitors probing the S2 active site pocket. Journal of Enzyme Inhibition and Medicinal Chemistry 34, 547-561. (doi:10.1080/14756366.2018.1564288) PMid:30696325
View Article PubMed/NCBIN. Aubin, E. Megnassan, Z. Nahossé, V. Frecer, N. Y. Thomas and S. Miertuš. 2017 Artemisinindipeptidyl vinyl sulfone hybrid inhibitors of Plasmodium falciparum falcipain 2 with favorable pharmacokinetic profile. J Drug Des Devel. 1, 11-28.
V. Frecer, M. Kabelác, P. D. Nardi, S. Pricl and S. Miertus. 2004 Structure-based design of inhibitors of NS3 serine protease of hepatitis C virus. J Mol Graph Model 22, 209-20. (doi:10.1016/S1093-3263(03)00161-X) 00161-X
View ArticleV. Frecer, E. Burello and S. Miertus. 2005 Combinatorial design of nonsymmetrical cyclic urea inhibitors of aspartic protease of HIV-1. Bioorg Med Chem. 13, 5492-501. (doi:10.1016/j.bmc.2005.06.026) PMid:16054372
View Article PubMed/NCBIB. Dali, M. Keita, E. Megnassan, V. Frecer and S. Miertuš. 2012 Insight into selectivity of peptidomimetic inhibitors with modified statine core for plasmepsin II of Plasmodium falciparum over human cathepsin D. Chem Biol Drug Des. 79, 411-30. (doi:10.1111/j.1747-0285.2011.01276.x) PMid:22129033
View Article PubMed/NCBIE. Megnassan, M. Keita, C. Bieri, A. Esmel, V. Frecer and S. Miertuš. 2012 Design of novel dihydroxynaphthoic acid inhibitors of Plasmodium falciparum lactate dehydrogenase. Med Chem. 8, 970-84. (doi:10.2174/157340612802084324) PMid:22741776
View Article PubMed/NCBIL. C. O. Owono, M. Keita, E. Megnassan, V. Frecer and S. Miertus. 2013 Design of thymidine analogues targeting thymidilate kinase of Mycobacterium tuberculosis. Tuberc Res Treat. 2013, 670836. (doi:10.1155/2013/670836) PMid:23634301
View Article PubMed/NCBIV. Frecer, F. Berti, F. Benedetti and S. Miertuš. 2008 Design of peptidomimetic inhibitors of aspartic protease of HIV-1 containing -Phe Psi Pro- core and displaying favourable ADME-related properties. J Mol Graph Model 27, 376-87. (doi:10.1016/j.jmgm.2008.06.006) PMid:18678515
View Article PubMed/NCBIV. Frecer, P. Seneci and S. Miertus. 2011 Computer-assisted combinatorial design of bicyclic thymidine analogs as inhibitors of Mycobacterium tuberculosis thymidine monophosphate kinase. J Comput Aided Mol Des. 25, 31-49. (doi:10.1007/s10822-010-9399-4) PMid:21082329
View Article PubMed/NCBIL. C. Owono Owono, F. Ntie-Kang, M. Keita, E. Megnassan, V. Frecer and S. Miertuš. 2015 Virtually designed triclosan-based inhibitors of enoyl-acyl carrier protein reductase of Mycobacterium tuberculosis and of Plasmodium falciparum. Mol Inform. 34, 292-307. (doi:10.1002/minf.201400141) PMid:27490275
View Article PubMed/NCBIA. Esmel, M. Keita, E. Megnassan, B. Toï, V. Frecer and S. Miertuš. 2017 Insight into binding mode of nitrile inhibitors of Plasmodium falciparum Falcipain-3, QSAR and Pharmacophore models, virtual design of new analogues with favorable pharmacokinetic profiles. J Comput Chem Molec Model 2, 103-24. (doi:10.25177/JCMP.2.1.5)
View ArticleY. K. H. Fagnidi, B. Toi, E. Megnassan, V. Frecer and S. Miertuš. 2018 In silico design of Plasmodium falciparum cysteine protease falcipain 2 inhibitors with favorable pharmacokinetic profile. J Anal Pharm Res. 7, 298-309. (doi:10.15406/japlr.2018.07.00244)
View ArticleInsight-II and Discover molecular modeling and simulation package, Version 2005. 2005 CA: Accelrys, Inc, San Diego.
W. Rocchia, S. Sridharan, A. Nicholls, E. Alexov, A. Chiabrera and B. Honig. 2002 Rapid grid-based construction of the molecular surface and the use of induced surface charge to calculate reaction field energies: applications to the molecular systems and geometric objects. J Comput Chem. 23, 28-37. (doi:10.1002/jcc.1161) PMid:11913378
View Article PubMed/NCBIM. K. Gilson and B. Honig. 1991 The inclusion of electrostatic hydration energies in molecular mechanics calculations. J Comput Aided Mol Des. 5,5-20. (doi:10.1007/BF00173467) PMid:2072125
View Article PubMed/NCBIDiscovery Studio molecular modeling and simulation program. Version 2.5. 2009 CA: Accelrys, Inc., San Diego.
S. Miertuš, E. Scrocco and J. Tomasi. 1981 Electrostatic interaction of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects. Chem Phys. 55, 117-129. (doi:10.1016/0301-0104(81)85090-2) 85090-2
View ArticleS. Miertuš and J. Tomasi. 1982 Approximate evaluations of the electrostatic free energy and internal energy changes in solution processes. Chem Phys. 65, 239-245. (doi:10.1016/0301-0104(82)85072-6) 85072-6
View ArticleV. Frecer and S. Miertuš. 1992 Polarizable continuum model of solvation for biopolymers. Int J Quant Chem. 42, 1449-1468. (doi:10.1002/qua.560420520)
View ArticleH. Li, J. Sutter and R. Hoffmann. 2000 HypoGen: an automated system for generating 3D predictive pharmacophore models. Güner OF, editor. 171-189.
QikProp Version 3.7, Release 14. 2014 NY: X Schrodinger, LLC, New York.
W. L. Jorgensen and E. M. Duffy. 2000 Prediction of drug solubility from Monte Carlo simulations. Bioorg Med Chem Lett. 10, 1155-8. (doi:10.1016/s0960-894x(00)00172-4) 00172-4
View ArticleW. L. Jorgensen and E. M. Duffy. 2002 Prediction of drug solubility from structure. Adv Drug Deliv Rev. 54, 355-66. (doi:10.1016/s0169-409x(02)00008-x) 00008-X
View ArticleE. Duffy and W. Jorgensen. 2000 Prediction of properties from simulations: free energies of solvation in hexadecane, octanol, and water. J Am Chem Soc. 122, 78-88. (doi:10.1021/ja993663t)
View ArticleP. Willett. 1994 Molecular Similarity in Drug Design. Dean PM, (Ed.) 110-137.
View ArticleA. M. Sapse, B. S. Schweitzer, A. P. Dicker, J. R. Bertino, and V. Frecer. 1992 Ab initio studies of aromatic-aromatic and aromatic-polar interactions in the binding of substrate and inhibitor to dihydrofolate reductase. Int. J. Pept. Prot. Res. 39, 18-23. (doi:10.1111/j.1399-3011.1992.tb01550.x) PMid:1299221
View Article PubMed/NCBIC. Lipinski, F. Lombardo, B. Dominy and P. Feeney. 2001 Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 46, 3-26. (doi:10.1016/s0169-409x(00)00129-0)