Muhammad Shoaib, Usman Ali
Email: usmanqureshi320@gmail.com, usmanqureshi@iccas.ac.cn (U. Ali), mshoaib285@gmail.com (M. Shoaib)
© 2019 Sift Desk Journals. All Rights Reserved
VOLUME: 4 ISSUE: 3
Muhammad Shoaib, Usman Ali
Email: usmanqureshi320@gmail.com, usmanqureshi@iccas.ac.cn (U. Ali), mshoaib285@gmail.com (M. Shoaib)
Zameer Hussain [1], Muhammad Shoaib*[1], Usman Ali *[1, 2], Hina Ramzan [1], Muhammad Ali [3], Amina Tariq [1], Muhammad Naqash [1]
[1] Department of Chemistry, University of Agriculture, Faisalabad, 38040, Pakistan.
[2] Beijing National Laboratories for Molecular sciences, Key Laboratory of Organic Solids, Institute of Chemistry Chinese Academy of Sciences, Beijing, 100190, People’s Republic of China.
[3]Department of Chemistry, Government College University, Faisalabad, 38040, Pakistan.
Nursel Acar(nursel.acar@ege.edu.tr)
Chunfu Shao(switeric@126.com)
Tifang Miao(miaotifang@163.com)
Koichi Kato(kato-k@kinjo-u.ac.jp)
Zameer Hussain, Muhammad Shoaib, Usman Ali, Hina Ramzan, Muhammad Ali, Amina Tariq, Mu-hammad Naqash , Theoretical study of Isoxazoles and their derivatives for evaluating its pharmaceuti-cal properties with density functional theory(2020)Journal of Computational Chemistry & Molecular Modeling 4(3) pp:415-426
Isoxazoles and their derivatives extracted from natural sources are limited in numbers. The isoxazole compounds and their derivatives have many importance in pharmaceutical industries like work as anti-tumor, anti-cancer, anti-bacterial, antifungal and antimicrobial agents. Therefore, this research work was focused to design the isoxazoles and their derivatives which have strong medicinal and pharmaceutical activities with computational chemistry. During this research work, UV/Visible absorption spectra of isoxazole and their derivatives were estimated for analyzing its absorption properties. Herein current simulation analysis different functionals of density functional theory like B3LYP, CAM-B3LYP, WB97XD and MPW1PW91 with 6-31G (d,p) basis set were employed for calculating energy gap (Eg) between highest occupied and lowest unoccupied molecular orbital states at ground state level. The calculated result reveals that our designed molecules have better pharmaceutical activities and can be use as anti-bacterial, antifungal and antimicrobial agents.
Isoxazole have condensed formula (C15H11NO) which is isomer of derivative of alpha pinene and also belong to class of ibotenic acid. All three atoms of this class are potentially reactive like chain of N-O-C. The danazol is the important interested compound which is analog of ethisteron due to its contraceptive activity [1].When alkenes and alkynes are corroded with the nitrate oxide by the process of 1,3 dipolar cyclo addition produced isoxazolines. In natural product range of synthesis the adaptable intermediate is formed which is known as 2,3 isoxazolines [ 4,5- dihydroisoxazoles] [2].The therapeutic category of “Anabolic” the compound 17- methylated – Andro-isoxazol (3, 2-c) isoxazole and 17 α -ethinyl-17β -hydroxy-androst -4- eno (2, 3-d) isoxazole belongs to class of antigonadotropin [3].The naphtha -[2, 3-d ] isoxazole-4, 9-di-ones explain as in situ generation of nitrite oxide necessary as primary material for synthesis of this compound but do not introduce the electron with drawing group (EWG) [4-6].The isoxazoles and difluoromethyl substituted pyrazoles synthesize by different method which is reported to their great importance and use of agriculture and medicinal industry [7].
A special type of isoxazole are synthesized which are very untreatable for chemists due to their biological activities are termed as fluorinated heterocyclic compound. A lot of examples are reported for the synthesis of trifluromethylated isoxazole but if we want to prepare the difluoromethyl then we must adopt the nucleophilic addition reaction [8, 9].
The isoxazole nucleus exist in various naturally obtained products and pharmacological characteristics have charmed the scientist form many years to discover and utilize isoxazole and its derivatives for medication purposes[10]. Various unique synthetic methods have been developed for the synthesis of isoxazole containing molecules. Its numerous derivatives have bundle of applications in the in the field of anti-HIV, photo -chemo-therapy, and anti-tumor [11].
In the various molecules which are biologically active compounds isoxazole basic nucleus is present such as ibotenic acid 1 and in various marketed medicines like Bextra and Valdecoxib[12]. In the field of the drug detection these compounds are being widely employed. Numerous methods are available for the preparation of the Isoxazoles like [3 + 2] cycloaddition of the alkyne with nitrile oxide. Traditional way of the synthesis of the nitrile oxide demands the de-hydration of the nitro-alkane[13-15]. The reaction of the aldoximes with the halogenating agents and oxidizing agents is also most widely being employed method for the preparation of the nitrile oxides [16, 17].
Various synthetic methods are also for the synthesis of isoxazole nucleus such as the cycloaddition reaction of the alkynes and alkenes with the nitrile oxides, and hydroxyl-amine reaction with the un-saturated ketones or 1, 3-di-ketones have resulted in the synthesis of the isoxazoles. Derivatives of the isoxazole have gained too much value in the medicinal and agricultural field.[5, 18].
Various naturally occurring compounds like muscimol , ibotenic acid, and a number of medicines just as cloxacillin , valdecoxib, and leflunomide contains isoxazole unit [9, 19-21]. The basic fuel conversion availabilities are the basic source of the thiophene, Furan, and pyrrole discharging into the environment. These are also present in numerous drugs; a number of models are available which predict the transport ability and the solubility of various medicines by recognizing the availability of the hydrogen bond locations. [22, 23].
Therefore, the inter-molecular hydrogen bonding among the H2O molecules and biologically important molecules is important for explaining the mechanism of reaction expressed by bio-chemical processes. Hydrogen bonding play key part in holding the inorganic matter like DNA and soft organic molecules and for the water molecules in the liquid state. Therefore, the existence of natural sources and all these types of conflicts can be avoided by employing isoxazole and its derivatives.
Scheme 1: Scheme of study of reference compound and their others derivative
Computational detail
The theoretical method had been employed for the evaluation of the hydrogen bonding between the oxazole, furan, water and isoxazole. For the optimization of heterocyclic single molecule dissolve in water and them adduct had been analyzed by DFT and method of Ab initio. Investigation of the Adduct formation for the basic compound had been employed with the help of various methods like MP2, VDZ, MP2, B3LYP functional with the 6-31+G basis set. Delocalization of the electron for the adducts had been done with the help of NBO method by using MP2 with 6-31G basis set.[22]
Newly synthesized compound was prepared from 3, 5 disubstituted pyrazoles and their carbonated derivatives were obtained from spirocyclic carbohydrates derivatives by intermediates of nitrile oxides. Examples were provided of isoxazol residues bonding through carbon linkage in which azoles part were attached to a carbohydrate. It was the synthesis of cluster of sugar chains contains complex carbohydrates microarrays had attached to a sugar platform that involve combination of C5, C6 ([24]. At last to optimize the poorly soluble drugs in straight series of 5-phenyl 3 isoxale 3carbonates were synthesized. Among the 4-position of substitution with polar functional group, it was unable to find out the strength, physicochemical properties. The use of capsaicin –stimulated CGRP was very efficient to analyze route of administration [25]
Method Selection
We can select the best Method of calculation for this purpose we can calculate the All methods energy value to the determent the reference valueMPW1PW91, CAM-B3LYP, B3LYP, W89BXD. The level of theory will be adopted by the measuring of the thermodynamic parameters for quantification with the help of B3LYP/6-31G(d) i.e. free energies (DG), relative energies (DE0), and enthalpies (DH). All the calculations were acceptable by using Gaussian Software03. With the help of density function theory and another theory like time dependent theory of DFT of software name as Gaussian 09 package which is used for calculate the geometry, electronic structure, and also the spectra of the given compound known as isoxazole and all other its derivatives. DFT is excellent and most useful method for describing organic compounds geometry. The chemical reagents and solvent used in the entire work are listed in table 1. All the chemical used in the research work were of analytical grades. And the software used for the computational study of isoxasoles derivatives are also described.
In the optimized structure of 3,5, diphenylisoxazole derivatives, the energy gap between HOMO and LUMO is 1.07 eV to 6.50 eV for MPW1PW91 functionals, 1.67 eV to 6.35 eV for B3LYP functionals and 0.21 eV to 7.75 eV for CAM-B3LYP functionals and 0.33 eV to 8.37 eV for WB97XD functionals. The complete detail of MPW1PW91 is present in (Table 3). If the HOMO to LUMO energy gap value of the compounds are less than 3eV it can be lies in the visible range whereas compounds having band gap more than 3 eV lies in the ultra-violet region.
By using density functional theory, the performance of different methods includes B3LYP, CAM-B3LYP, WB97XD and MPW1PW91. has been studied.
Frontier molecular orbitals
Twenty-five compounds of isoxazole derivatives were studied along with one reference compound with the help of density functional theory (DFT) and time dependent density functional theory (TD-DFT) at MPW1PW91/6-31 G (d, p) level of theory which is found to be more suitable.
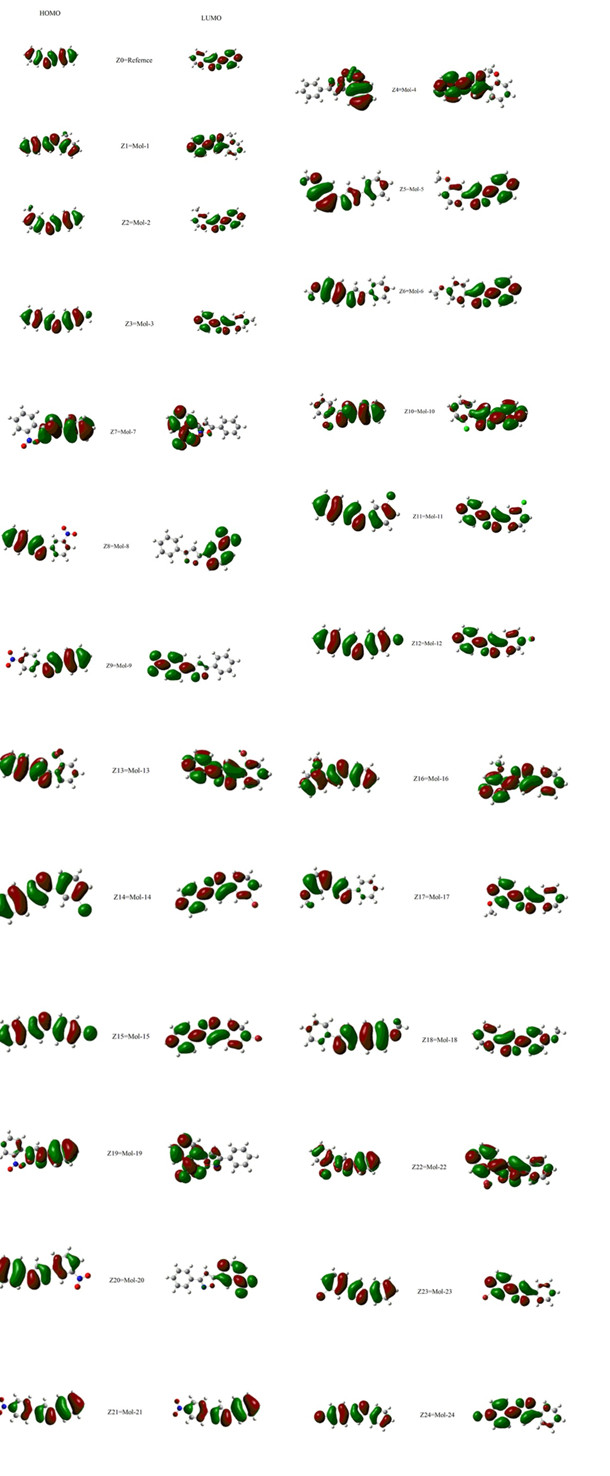
Figure. 1 The distribution pattern of HOMO and LUMO of (Ref., Mol-0 Mol-24) by using functional MPW1PW91 with 6-31/G (d, p)
In Mol-1and Mol-3 the Methyl group which is electron donating by hyper conjugation is substituted o
n benzene ring at o-position and p-position respectively it enhances the delocalization in case of HOMO but in case of LUMO the delocalization is less on that ring at which electron donating group is attached. In Mol-2 the Methyl group is substituted on benzene ring at m-position at which it is less effective the delocalization in case of HOMO but in case of LUMO the delocalization is more as compare to above because electron donating nature little effect at m-position.
In Mol-4,Mol-16 and Mol-6, Mol-18 the Methoxy group which is electron donating by Resonance is substituted on benzene ring at o-position and p-position respectively which “slightly enhance the delocalization character “but due to presence of oxygen the left sided ring shows no localization the attraction towards that ring at which MeO group attached in case of o-position but in case of p-position little localization on left benzene ring in case of HOMO but in case of LUMO the delocalization is so less on that ring at which electron donating group is attached. In Mol-5, Mol-17 MeO group is attached at m-position therefore it shows little effect as compare to above ones and left sided ring shows localization is more as compare to p-position in HOMO.
In Mol-7,Mol-19 and Mol-9,Mol-21 Nitro group is attached on benzene ring at o-position and p-position respectively which is electron with drawing due to Resonance and it reduce the localization of electron at that ring on which this group is attached and also isoxazole ring in case of HOMO but in case of LUMO there is no localization on right benzene ring and partially on isoxazole also due to Nitrogen which is electronegative. In Mol-8, Mol-20 the nitro group is attached at m-position which is not significantly contribute in electron delocalization in case of both HOMO and LUMO.
In Mol-10, Mol-13, Mol-22 and Mol-12, Mol-15, Mol-24 Halogen group is attached at o-position and p-position of benzene ring they show dual effect due to electronegativity (Inductive effect and electron donating by resonance). At o-position its resonance effect is prominent and halogen increase the localization shows in HOMO and in case of LUMO it has slight effects on electron delocalization as compare to HOMO. In case of p-position inductive effect is prominent cause less localization as compare to o-position. In Mol-11, Mol-14, Mol-23 the halogen group are present at m-position and its influence on HOMO and LUMO has not seen noticeably.
Table1. Oscillator strength (f), DE, molecular orbital contributions related to electronic transitions (ETC %), λ max values and theoretical λ max values calculated with MPW1PW91
|
Molecules |
Experimental Wavelength(nm) |
Theoretical Wavelength(nm) |
Δ E (eV) |
Oscillator strength(f) |
|
Assignment |
|
Reference
|
263 |
265.1 |
4.68 |
0.7077 |
|
H-0->L+0(+71%) |
|
Mol-1 |
_ |
262.4 |
4.72 |
0.8174 |
|
H-0->L+0(+63%) |
|
Mol-2 |
_ |
270.2 |
4.59 |
0.7273 |
|
H-1->L+0(+48%) |
|
Mol-3 |
_ |
266.5 |
4.65 |
0.5389 |
|
H-1->L+0(+94%) |
|
Mol-4 |
_ |
278.0 |
4.46 |
0.1285 |
|
H-0->L+0(+87%) |
|
Mol-5 |
_ |
281.3 |
4.41 |
0.0057 |
|
H-0->L+0(+88%) |
|
Mol-6 |
_ |
285.7 |
4.34 |
0.1152 |
|
H-0->L+0(+89%) |
|
Mol-7 |
_ |
337.4 |
3.68 |
0.0010 |
|
H-0->L+0(+97%) |
|
Mol-8 |
_ |
431.9 |
2.87 |
0.0127 |
|
H-0->L+0(+99%) |
|
Mol-9 |
_ |
339.1 |
3.66 |
0.0040 |
|
H-0->L+0(+99%) |
|
Mol-10 |
_ |
262.8 |
4.72 |
0.7111 |
|
H-0->L+0(+92%) |
|
Mol-11 |
_ |
268.6 |
4.62 |
0.5326 |
|
H-0->L+0(+82%) |
|
Mol-12 |
_ |
268.8 |
4.61 |
0.6693 |
|
H-0->L+0(+53%) |
|
Mol-13 |
_ |
266.6 |
4.65 |
0.6208 |
|
H-0->L+0(+84%) |
|
Mol-14 |
_ |
268.9 |
4.61 |
0.5229 |
|
H-0->L+0(+81%) |
|
Mol-15 |
_ |
269.7 |
4.60 |
0.7129 |
|
H-1->L+0(+63%) |
|
Mol-16 |
_ |
260.8 |
4.75 |
0.3322 |
|
H-0->L+0(+59%) |
|
Mol-17 |
_ |
287.7 |
4.31 |
0.2986 |
|
H-0->L+0(+87%) |
|
Mol-18 |
_ |
284.8 |
4.35 |
0.6726 |
|
H-0->L+0(+94%) |
|
Mol-19 |
_ |
330.0 |
3.76 |
0.0398 |
|
H-0->L+0(+79%) |
|
Mol-20 |
_ |
333.7 |
3.72 |
0.0445 |
|
H-0->L+0(+75%) |
|
Mol-21 |
_ |
330.0 |
3.76 |
0.2597 |
|
H-0->L+0(+98%) |
|
Mol-22 |
_ |
257.5 |
4.81 |
0.2012 |
|
H-1->L+0(+50%) |
|
Mol-23 |
_ |
269.4 |
4.60 |
0.4572 |
|
H-1->L+0(+44%) |
|
Mol-24 |
_ |
274.9 |
4.51 |
0.9772 |
|
H-0->L+0(+86%) |
Table 2. Dipole moment of excited and ground state with reference mol. (Z0 to Z24) with MPW1PW91/6-31 G (d, p)
|
Molecules |
µg |
µe |
µe-µg |
|
Reference |
2.817 |
4.010 |
1.193 |
|
Mol-1 |
2.605 |
3.651 |
1.046 |
|
Mol-2 |
3.712 |
5.293 |
1.581 |
|
Mol-3 |
2.829 |
4.083 |
1.254 |
|
Mol-4 |
3.819 |
5.182 |
1.363 |
|
Mol-5 |
2.862 |
3.975 |
1.113 |
|
Mol-6 |
1.656 |
2.483 |
0.827 |
|
Mol-7 |
5.835 |
7.771 |
1.936 |
|
Mol-8 |
3.547 |
4.502 |
0.955 |
|
Mol-9 |
6.605 |
7.909 |
1.304 |
|
Mol-10 |
4.011 |
5.621 |
1.610 |
|
Mol-11 |
2.098 |
2.686 |
0.768 |
|
Mol-12 |
3.755 |
4.905 |
1.150 |
|
Mol-13 |
1.982 |
2.752 |
0.770 |
|
Mol-14 |
2.106 |
2.724 |
0.618 |
|
Mol-15 |
3.698 |
4.848 |
1.150 |
|
Mol-16 |
2.406 |
3.528 |
1.122 |
|
Mol-17 |
1.441 |
2.323 |
0.882 |
|
Mol-18 |
4.472 |
6.049 |
1.577 |
|
Mol-19 |
6.121 |
8.321 |
2.200 |
|
Mol-20 |
6.538 |
9.228 |
2.690 |
|
Mol-21 |
4.975 |
6.098 |
1.123 |
|
Mol-22 |
3.907 |
5.594 |
1.687 |
|
Mol-23 |
3.928 |
5.529 |
1.601 |
|
Mol-24 |
2.565 |
3.677 |
1.112 |
Dipole Moment
It is the property of molecule which tells us about the nature of the molecule and its solubility. After calculating dipole moment, we concluded that the solubility of the reference molecules and its derivatives give maximum % yield in Water as compare to Benzene, Chloroform, Ethanol, n.hexane. Table 4.2 shows all the dipole moment value of reference compound and their derivatives.
Table 3. HOMO, LUMO and Eg values reference mol. (Z0 to Z24) with MPW1PW91/6-31 G (d,p)
|
Molecules |
Homo (eV) |
Lumo (eV) |
Band Gape(eV) |
|
Reference |
6.50162 |
1.07757 |
5.42 |
|
Mol-1 |
6.51087 |
1.24764 |
5.26 |
|
Mol-2 |
6.57019 |
1.20329 |
5.37 |
|
Mol-3 |
6.38026 |
1.40846 |
4.97 |
|
Mol-4 |
6.32366 |
1.20302 |
5.12 |
|
Mol-5 |
6.23495 |
1.18587 |
5.05 |
|
Mol-6 |
6.05644 |
1.15104 |
4.91 |
|
Mol-7 |
6.67033 |
2.05936 |
4.61 |
|
Mol-8 |
6.81863 |
2.36521 |
4.45 |
|
Mol-9 |
6.89238 |
2.45801 |
4.43 |
|
Mol-10 |
6.58162 |
1.26370 |
5.32 |
|
Mol-11 |
6.64040 |
1.40737 |
5.23 |
|
Mol-12 |
6.60230 |
1.40030 |
5.20 |
|
Mol-13 |
6.55958 |
1.27377 |
5.29 |
|
Mol-14 |
6.63060 |
1.40792 |
5.22 |
|
Mol-15 |
6.57944 |
1.40601 |
5.17 |
|
Mol-16 |
6.51006 |
1.04900 |
5.46 |
|
Mol-17 |
6.27903 |
1.22506 |
5.05 |
|
Mol-18 |
6.05889 |
1.01934 |
5.04 |
|
Mol-19 |
6.78870 |
2.31433 |
4.47 |
|
Mol-20 |
6.89891 |
2.44250 |
4.46 |
|
Mol-21 |
6.96966 |
2.65828 |
4.31 |
|
Mol-22 |
6.67087 |
1.22342 |
5.45 |
|
Mol-23 |
6.68230 |
1.50533 |
5.18 |
|
Mol-24 |
6.56339 |
1.49336 |
5.07 |
Absorption Spectra:
In Table 1 Mol-1, Mol-10, Mol6, Mol-22, the value of absorption spectra is 262.4, 262.8, 260.8, 257.5, nm respectively which is less as compare to reference value 265.1nm because the left-hand benzene ring is substituted electron donating group at o- position which increase he energy and when energy is increase wave length is decreased.
In Mol-3 the absorption value is 266.5 nm because methyl group is attached to benzene ring at p-position which is less effective as compare to o-position, so value is slightly increase.
In Mol-13 the absorption value is 265.1 nm because halogen group is attached to benzene ring at o-position which shows dual effect in this resonance effect is less, so value is slightly decrease.
In Mol-2 the absorption value is 270.4 nm because methyl group is attached to benzene ring at m-position which does not shows electron donating nature, so value is higher as compare to reference value.
In Mol-11, Mol-12, Mol-14, Mol-15, Mol-23, the value of absorption spectra is, 268.6, 268.8, 268.9,269.7, 269.4 nm respectively which is more as compare to reference value 265.1nm because the benzene ring is substituted halogen group gat o- position and p- position which shows resonance effect very little and increase the energy which decrease the wave length.
In Mol-5, Mol-6, Mol-17, Mol-18 the value of absorption spectra is, 281.3, 285.7, 287.7, 284.8, nm respectively which is more as compare to reference value 265.1nm because the benzene ring is substituted methoxy group at o- position and p- position which shows electron donating effect very little but oxygen is more electro negative and reduce the energy of that ring which cause increase the wave length.
In Mol-4, Mol-16, the value of absorption spectra is, 278.0, 260.8 nm respectively the methoxy group is attached near the nitrogen and oxygen atom which is electro negative group which hindered the electron donating nature of methoxy and value of absorption is less as compare to its other position.
In Mol-7, Mol-8, Mol-9, Mol-17, Mol-18, Mol-19, the value of absorption spectra is 337.4,431.9, 339.1, 330.0, 333.7, 330.0 nm respectively which is more as compare to reference value 265.1nm because the benzene ring is substituted electron withdrawing group at o-p- position and also m-position which decrease the energy and when energy is decrease wave length is increased. In Fig 2 All the graph shows the supporting material of above discussion their peaks are represent the absorption value and their difference with respect to reference value.
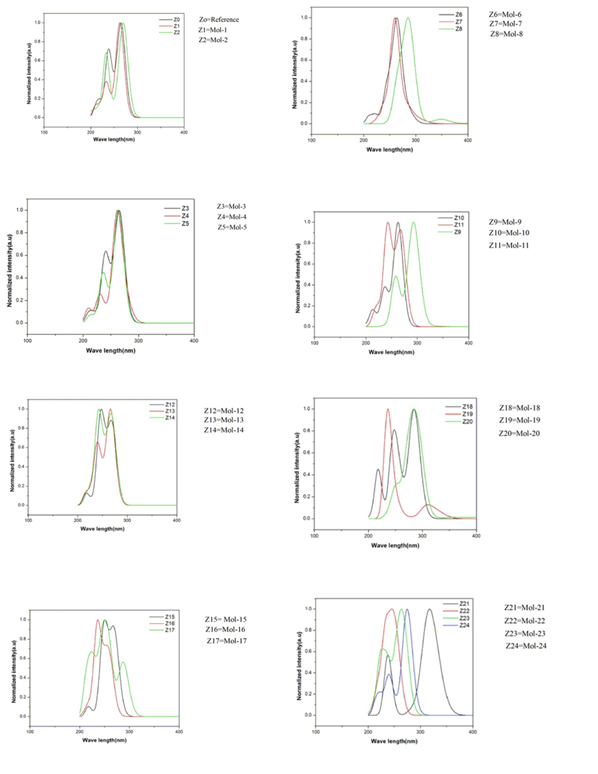
Figure.2 Absorption spectra of 3,5-diphenylisoxazol derivatives by the comparison of all the molecules with the Ref. molecule (Z0 to Z24) by using MPW1PW91 with 6-31/G (d, p)
First of all, TD-CAM-B3LYP absorption wavelength of 3,5-diphenylisoxazol derivatives are studied. This study is done by taking the peak position in consideration obtained from the absorption spectra. The absorption spectra of Mol-12 show the value of λmax which have very close resemblance to the experimental data in this literature it shows only one broad band at 337 nm while the absorption spectra of Mol-7reveal the value which is highly overestimated to the data in this literature. In the absorption spectra calculated with TD-CAM-B3LYP by analyzing the peak position it was revealed that the Mol-6, Mol-10, Mol-11, Mol-14 to Mol-16, Mol-18 to Mol-20, Mol-22 and Mol-23 have two distinct absorption broad band in the absorption range of 214 nm to 387 nm. Mol-7 and Mol-8 also have two distinct absorption bands in the range of 303 nm to 437 nm. The Mol-5, Mol-9, Mol-13, Mol-17 and Mol-21 can show only one strong broad band in the absorption range of 259 nm to 345 nm.
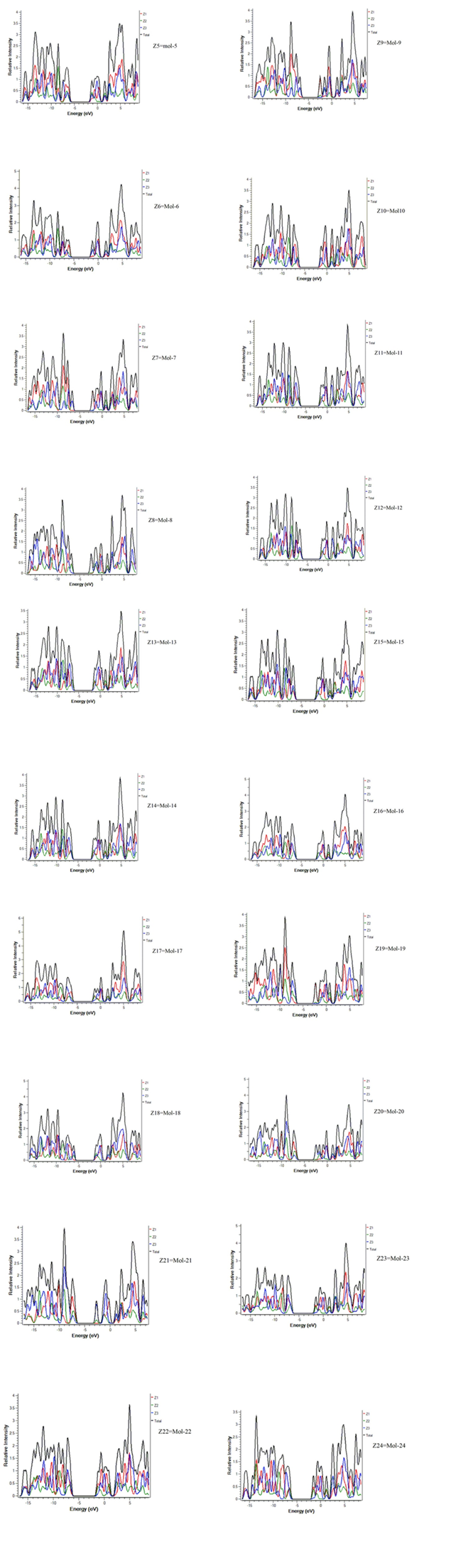
Figure. 3 Mullikan spectra of 3,5-diphenylisoxazol derivatives by the comparison of all the molecules with the Ref. molecule (Z0 to Z24) by using MPW1PW91 with 6-31/G (d, p)
Density of State
The study of frontier molecular orbitals can also be done with this functional for knowing the best functional for 3,5-diphenylisoxazol derivatives. The following are the images of HOMO LUMO orbitals of 3,5-diphenylisoxazol derivatives with WB97XD functional Reported value of 3,5-diphenylisoxazol =263 nm, B3LYP gives =286.41nm. ,CAM-B3LYP=249.80nm., and MPW1PW91=265.1n.m . There for MPW1PW91 is the best method for Calculation and determination of 3,5-diphenylisoxazol derivatives value because it is close to the reported experimental wave length value. In the fig. 3 the Z1 is shows the left side of the benzene ring, Z2 represent the central isoxazole ring and the Z3 is the last and right benzene ring.
Isoxazole contain fascinating chemistry. In the isoxazole 3,5 diphenylisoxazole was the basic part of their structure. They are generally distributed in the nature which are generally found in bacteria and plants and hence to living systems. The following results shown by the computational study of structure and optical properties like UV/Vis spectra of isoxazole derivatives using density functional theory and time dependent density functional theory methods were used.
A series of 3,5 diphenylisoxazole derivatives were experienced for the explanation of best methodology for the forecast of UV/Visible spectra and MPW1PW91 functional was gave the satisfactory result.
In the graphical comparison the result was revealed that the functionals CAM-B3LYP and MPW1PW91 gave the close calculated λ max value when compared with the experimental λ max value. The functional B3LYP gave the over assessed λ max value but CAM-B3LYP and WB97XD gave the under assessed λ max value in comparison with experimental value. The results clear that the method CAM-B3LYP was the best method for the 3,5 diphenylisoxazole derivatives. WB97XD also give the best result but not like CAMB3LYP.
The Overall calculated result reveals that our designed molecules have better pharmaceutical activities and can be use as anti-bacterial, antifungal and antimicrobial agents
Hernández-Linares, M.G., et al., Stereospecific synthesis of new steroidal isoxazoles in dry media. Steroids, 2011. 76(14): p. 1521-1526. PMid:21872615
View Article PubMed/NCBIKarthikeyan, K., et al., Synthesis and antinociceptive activity of pyrazolyl isoxazolines and pyrazolyl isoxazoles. Bioorganic & medicinal chemistry letters, 2009. 19(13): p. 3370-3373. PMid:19481931
View Article PubMed/NCBIParr, M.K., et al., Identification of steroid isoxazole isomers marketed as designer supplement. Steroids, 2009. 74(3): p. 322-328. PMid:19061909
View Article PubMed/NCBISantos, M.M., et al., Reaction of naphthoquinones with substituted nitromethanes. Facile synthesis and antifungal activity of naphtho [2, 3-d] isoxazole-4, 9-diones. Bioorganic & medicinal chemistry letters, 2010. 20(1): p. 193-195. PMid:19926280
View Article PubMed/NCBISaeed, U., et al., Designation and Match of Non‐Fullerene Acceptors with X‐Shaped Donors toward Organic Solar Cells. ChemistrySelect, 2019. 4(13): p. 3654-3664.
View ArticleAli, U., et al., Molecular designing of naphthalene diimide based fullerene-free small organic solar cell-Acceptors with high photovoltaic performance by density functional theory. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 2020. 228: p. 117685. PMid:31748156
View Article PubMed/NCBIYang, X., et al., Synthesis of bromodifluoromethyl substituted pyrazoles and isoxazoles. Journal of Fluorine Chemistry, 2010. 131(3): p. 426-432.
View ArticleYang, X., et al., Regioselective nucleophilic addition of 3-aryl-5-difluoromethyl-isoxazoles to aldehydes. Journal of Fluorine Chemistry, 2013. 145: p. 1-7.
View ArticleShoaib, M., et al., Theoretical Investigation of Perylene Diimide derivatives as Acceptors to Match with Benzodithiophene based Donors for Organic Photovoltaic Devices. Zeitschrift für Physikalische Chemie.
Allen, T.M. and P.R. Cullis, Drug delivery systems: entering the mainstream. Science, 2004. 303(5665): p. 1818-1822. PMid:15031496
View Article PubMed/NCBITripathi, V.K., et al., Synthesis and biological evaluation of novel isoxazoles and triazoles linked 6-hydroxycoumarin as potent cytotoxic agents. Bioorganic & medicinal chemistry letters, 2014. 24(17): p. 4243-4246. PMid:25088398
View Article PubMed/NCBIAl-Sehemi, A.G., A. Irfan, and A.M. El-Agrody, Synthesis, characterization and DFT study of 4H-benzo [h] chromene derivatives. Journal of Molecular Structure, 2012. 1018: p. 171-175.
View ArticleCho, H.-S., et al., Effects of enzymatic hydrolysis on lipid extraction from Chlorella vulgaris. Renewable Energy, 2013. 54: p. 156-160.
View ArticleChoi, W.Y. and H.Y. Lee, Effective production of bioenergy from marine Chlorella sp. by high-pressure homogenization. Biotechnology & Biotechnological Equipment, 2016. 30(1): p. 81-89.
View ArticleColla, L.M., T.E. Bertolin, and J.A.V. Costa, Fatty acids profile of Spirulina platensis grown under different temperatures and nitrogen concentrations. Zeitschrift für Naturforschung C, 2004. 59(1-2): p. 55-59. PMid:15018053
View Article PubMed/NCBIPage, L.W., et al., The acid-mediated intramolecular 1, 3-dipolar cycloaddition of derived 2-nitro-1, 1-ethenediamines for the synthesis of novel fused bicyclic isoxazoles. Tetrahedron Letters, 2010. 51(26): p. 3388-3391.
View ArticleAli, U., et al., Designing difluoro substituted benzene ring based fullerene free acceptors for small Naphthalene Di-Imide based molecules with DFT approaches. Optical and Quantum Electronics, 2019. 51(10): p. 332.
View ArticleRajawinslin, R., et al., Selectfluor mediated one pot synthesis of cyclohexanone ring fused isoxazole derivatives. Tetrahedron, 2014. 70(41): p. 7505-7510.
View ArticleSinghal, A., et al., Hypervalent iodine mediated synthesis of di-and tri-substituted isoxazoles via [3+ 2] cycloaddition of nitrile oxides. Tetrahedron Letters, 2016. 57(7): p. 719-722.
View ArticleAli, U., et al., Benchmark study of benzamide derivatives and four novel theoretically designed (L1, L2, L3, and L4) ligands and evaluation of their biological properties by DFT approaches. Journal of molecular modeling, 2019. 25(8): p. 223. PMid:31302811
View Article PubMed/NCBIAjmal, M., et al., Designing indaceno thiophene-based three new molecules containing non-fullerene acceptors as strong electron withdrawing groups with DFT approaches. Journal of molecular modeling, 2019. 25(10): p. 311. PMid:31512040
View Article PubMed/NCBIKaur, D. and S. Khanna, Intermolecular hydrogen bonding interactions of furan, isoxazole and oxazole with water. Computational and Theoretical Chemistry, 2011. 963(1): p. 71-75.
View ArticleWaqas, A., et al., Substitutional effect of different bridging groups on optical and charge transfer properties of small bipolar molecules for OLEDs. Journal of Physical Organic Chemistry, 2019. 32(11): p. e4000.
View ArticleLugiņina, J., et al., A concise synthesis of sugar isoxazole conjugates. Tetrahedron Letters, 2013. 54(39): p. 5328-5331.
View ArticleRatcliffe, P., et al., Discovery of potent, soluble and orally active TRPV1 antagonists. Structure-activity relationships of a series of isoxazoles. Bioorganic & medicinal chemistry letters, 2011. 21(15): p. 4652-4657. PMid:21723725
View Article PubMed/NCBI