Covadonga R. Arias
Phone: 1-334-844-9215, Fax: 1-334-844-9208;
E-mail: ariascr@auburn.edu
© 2019 Sift Desk Journals. All Rights Reserved
VOLUME: 2 ISSUE: 2
Page No: 138-154
L. Raya,b, W. Caia, E. Willmona, c, and C. R. Ariasa
Jovanka Lukic(lukicjovanka@imgge.bg.ac.rs)
Hanguo Xiong(xionghanguo@163.com)
Qingyun Yan(yanqy6@mail.sysu.edu.cn)
Sajjad Pourmozaffar(Sajjad5550@gmail.com)
Cova Arias et al., Fish are not alone: characterization of the gut and skin microbiomes of Largemouth Bass (Micropterus salmoides), Bluegill (Lepomis macrochirus), and Spotted Gar (Lepisosteus oculatus)(2019)SDRP Journal of Aquaculture, Fisheries & Fish Science 2(2)
The objective of this study was to characterize the gut and skin microbiomes of three common freshwater fishes including two important sport fishes, Largemouth Bass Micropterus salmoides and Bluegill Lepomis macrochirus as well as the distantly-related Spotted Gar Lepisosteus oculatus. Skin and gut samples were collected in August and November 2014, and May 2015. All samples were sequenced as paired-end reads of the 16S rRNA gene via the Illumina MiSeq platform. More than 5M reads were analyzed representing 4,130 and 2,744 OTUs from gut and skin samples, respectively. Approximately 51.84% of the total OTUs were shared between the skin and gut bacterial communities. Good’s coverage was higher than 98% in all samples. Spotted Gar exhibited the most diverse skin microbiome, while Largemouth Bass was the least diverse species in terms of both the skin and gut microbiome compositions. The highest diversity in the gut microbiome was observed in Bluegill; however, the bacterial communities of Spotted Gar were the most variable across seasons. Seasonal changes in bacterial community structures were also observed. For both the skin and the gut microbiomes, sampling date was found to exert a stronger influence on microbial composition than the species itself; however, season had a lesser impact on the gut microbiome than in the skin indicating the gut microbiomes are more stable. This study provides baseline data on the bacterial symbiont communities of three iconic freshwater fish species in North America. Our data could be used in future studies to identify environmental stressors that result unbalanced microbiomes and loss of homeostasis.
Keywords: microbiome, Largemouth bass, Bluegill, Spotted Gar, Alabama
The term microbiome was first coined by Lederberg and McCray (2001) to describe the ecology of commensal, symbiotic, and pathogenic microbes that share space on and within a host. For over a century, health professionals have associated microorganisms with disease-causing agents, but recent studies have proven that the majority of microbes associated with our external and internal surfaces are indeed symbiotic in nature and some are key contributors to host homeostasis (Backhed et al., 2005; Hooper, 2009; Larsen et al., 2013). In order to understand disease predispositions and pathogenesis, it is first important to better understand the “normal” or healthy states and functions of the microbiome (Turnbaugh et al., 2007). In doing so, it is imperative not only to characterize the microbiome, but also to learn about the factors that influence the distribution and evolution of these microbial symbionts.
Earlier studies on bacterial communities in fish suggested a passive colonization of the host by microbes found in the surrounding environment (Horsley, 1977). Those studies were largely biased due to the use of culture-dependent methods that vastly underestimated the species richness of the microbial communities. With advancements in next generation sequencing technologies, exploring the richness of fish microbiomes became attainable. Recent studies showed that, despite the dynamic nature of these microbial communities and the intrinsic variation found within species, fish microbiomes displayed a marked species-specificity suggesting a long story of coevolution between fish host and microbes (Larsen et al., 2013; Chiarello et al., 2015). The body of current literature is largely biased towards studies on gut microbiome of aquacultured fishes because of its anticipated importance in digestion, disease control, and overall health (Huber et al., 2004). However, some studies indicate that rearing or holding animals under captive conditions alters the microbiome of the host (Dhanasiri et al., 2010; Hird, 2017). This may hinder or distort observations of natural variation, thereby limiting evolutionary analyses of a host’s microbiome. Therefore, to truly understand the complex dynamics between fish and their microbiomes, more studies examining the microbial communities in wild fishes are needed since much is still unknown about the composition and structural function of these communities (Clements et al., 2007; Nayak, 2010).
The gut microbiome is now credited for playing important roles in the development of host immune functions, epithelial renewal, nutrition, digestive processes, and xenobiotic metabolism (Uchii et al., 2006; Nayak, 2010; Mouchet et al., 2011; Llewellyn et al., 2014). An integrative system for host defense against disease is posit as a symbiotic partnership between the host’s gut epithelium, immune system, and the commensal gut communities (Llewellyn et al., 2014). This line of defense helps make up the gut’s immune system, more commonly referred to as the gut-associated lymphoid tissues (GALT), which defends the host against invading pathogens and regulates the immune system of the digestive tract (Rhee et al., 2004; Nayak, 2010). Thus, the gut microbiome helps defend the host, not only by educating and boosting the immune system, but also by enhancing it via inhibition of invading pathogens by either competitive exclusion or by the production of toxic secondary metabolites that prevent colonization (Wells et al., 1988; Balcazar et al., 2006; Llewellyn et al., 2014). Therefore, disruption of these important commensal communities can lead to a “dysbiotic” state (Nayak, 2010; Llewellyn et al., 2014). Once the balance of the gut microbiome is disturbed, potentially pathogenic transient microbes may colonize the gut resulting in disease for the host. Although used frequently in the literature concerning disease, the term dysbiosis is still somewhat subjective since the “natural” or normal gut microbiota is largely undefined for most host species.
Similarly, the skin microbiome, in conjunction with the skin and mucosal epithelia, serves as the first line of defense providing both a physical and chemical barrier against invading pathogens (Ellis, 1999; Esteban, 2012; Peatman et al., 2015). The mucosal surface along with its commensal bacterial communities helps provide protection by inhibiting the attachment, invasion, and growth of foreign bacteria on or within host tissues. Although less studied than the gut microbiome, the establishment and composition of the microbiome denotes a complex coevolution between the host and its microbial partners which has resulted in a relatively stable and mutually beneficial relationship between the two (Peatman et al., 2015). Experimental studies have shown that disrupting the skin & mucus microbiomes of fishes led to an increased host susceptibility to bacterial infections and disease (Cipriano and Dove, 2009; Littman and Pamer, 2011; Mohammed and Arias, 2015; Lockesh and Kiron, 2016).
The overall goal of this study was to expand our current knowledge on fish microbiomes to include the skin and gut microbiomes of important freshwater sport fishes. Our working hypothesis was that fish species would exert the highest influence on microbiome composition particularly in more stable gut communities. Specific objectives aimed at i) characterizing the gut and skin microbiomes of three common freshwater fishes in the Southeast of the USA, including two commercially valuable sport fishes, Largemouth Bass Micropterus salmoides and Bluegill Lepomis macrochirus as well as the more primitive Spotted Gar Lepisosteus oculatus, and ii) to identify potential seasonal effects on the core communities.
Sample collection. Fish were collected from the Lake Guntersville reservoir on the Tennessee River in northern Alabama (34.4054° N, 86.1984° W). Sample collections took place in August and November 2014 and in May 2015. Fishes were captured via electrofishing techniques (7.5 GPP Smith Root electrofishing boat) and maintained in live-wells until they could be returned to the shoreline for harvesting and preservation of target tissues. Four individuals per species were collected during each sampling event. Average length (mm±SD) was 180±34, 558±89, and 367±47 for Bluegill, Spotted Gar, and Largemouth Bass, respectively. Upon arrival to the docking site, fishes were humanely euthanized according to Auburn University-approved IACUC (Institutional Animal Care and Use Committee) protocols, segregated by species, and placed on ice for immediate processing of tissues. A fin clip of the dorsal fin of approximately 1 cm2 was aseptically removed and fixed in RNAlater® (Thermo Fisher Scientific, Waltham, MA). The ventral and lateral sides of the fish, were sprayed with 70% isopropanol and wiped clean repeatedly (x3) in effort to eliminate the contamination of gut microbiome samples with remnant communities from the external surfaces. Once excess isopropanol had been sufficiently dried from the skin, fish were first squeezed in an anterior to posterior direction to allow gut contents to be expelled from the urogenital pore onto a sterilized spatula. Fecal contents were fixed in RNAlater®. Samples were held at 4°C for a minimum of 6 hours to allow for thorough penetration of the preservation media into tissues. Samples were then transferred to -80°C storage until DNA extractions were performed.
DNA extraction, PCR Amplification, and Sequencing. Samples were processed as previously described (Larsen et al., 2013; Larsen et al., 2014b). Briefly, DNA from fin clips was extracted using the DNeasy® Blood & Tissue Kit (Qiagen, Valencia, CA; tissue kit), following manufacturer’s instructions, including pre-treatment steps to lyse Gram-positive bacteria. DNA from fecal samples (180-200 mg) was extracted using the QIAmp® DNA Stool Mini Kit (Qiagen, Valencia, CA; stool kit). Total DNA concentrations were quantified using a NanoDrop ND-1000 spectrophotometer (Thermo Scientific, Nanodrop Technologies, Wilmington, DE, USA). A total of 36 skin DNA samples were extracted (3 species x 4 replicates x 3 sampling events) while only 35 DNA samples from fecal matter were available. One of the Spotted Gar individuals collected in November did not yield sufficient fecal content for extraction. For consistency, the skin sample from that individual was also eliminated from the study.
A total of 70 (35 skin and 35 gut) samples were submitted to MR DNA® (Shallowater, TX, USA) for PCR amplification followed by sequencing using the Illumina MiSeq platform. Universal bacterial primers 515 F (5'-GTGCCAGCMGCCGCGGTAA-3') and 806R (5'-GGACTACHVGGGTWTCTAAT-3') with a barcode on the forward primer were used to target the 16S rRNA gene V4 variable region. The HotStarTaq Plus Master Mix Kit (Qiagen, USA) was used to run all samples under the following PCR conditions: an initial denaturation step for 3 minutes at 94°C followed by 28 cycles of 94°C for 30 s (denaturing), 53°C for 40 s (annealing), and 72°C for 1 min (extension) before performing a final elongation step for 5 min at 72°C. Following amplification, PCR products for all samples were run through a 2% agarose gel to verify successful amplification and relative band intensity of the target DNA. Multiple samples were pooled together and purified using calibrated Ampure XP beads to prepare the Illumina DNA library prior to sequencing. All samples were sequenced as paired-end reads on the Illumina MiSeq platform following the manufacturer’s instructions. Resulting sequences were processed using a proprietary pipeline (MR DNA, Shallowater, TX, USA). Sequencing data were joined, and all barcodes, primers, and sequences <150 bp were removed. Additionally, sequences with ambiguous base calls and spans of identical monomer units longer than 6 bp were removed. Denoising of sequences was also performed, and operational taxonomic units (OTUs) were generated. Cut-offs for OTU assignment were defined at a 97% similarity (<3% sequence variation) in concurrence with the prokaryotic species concept (Rosello-Mora, 2005). Taxonomic classifications were obtained using BLASTN against the GreenGenes database (DeSantis et al., 2006).
Data analysis. Sequences were randomly selected from each sample in order to standardize sampling effort to that of the sample that returned the least number of sequences (N=11,574 for skin and N=52,205 for gut samples). Mothur v.1.33.3 (Schloss et al., 2009) was used to generate rarefaction curves and to calculate diversity statistics including Good’s coverage, Shannon Evenness Index (SEI), abundance-based coverage estimation (ACE), Chao1, observed OTUs, and shared OTUs. SAS 9.2 (Statistical Analysis System, SAS Institute, Cary, NC) was used to run both one-way ANOVAs with Tukey multiple comparison tests (α = 0.05) as well as two-way ANOVAs in order to determine differences in the observed species richness (in observed OTUs), the total predicted species richness (ACE and Chao1), and species evenness (SEI) between samples. One-way ANOVAs were run first to determine potential differences between species and sampling dates, followed by two-ANOVAs to determine if a significant interaction variable existed. OTU tables including all samples were loaded into PRIMER v6 (Primer-E Ltd, Plymouth, UK) for clustering using the similarity matrix and analysis of similarities (ANOSIM). Genera tables were also loaded into PRIMER for similarity percentages (SIMPER) analysis in order to determine specific taxonomical differences between communities. The cut-off for low contributions was set to the default at 90%.
Sequencing of the 16S rRNA gene using the Illumina MiSeq platform resulted in a total of 5,419,640 sequences for all samples representing 4,527 total OTUs. Out of those, 2,744 OTUs were identified in skin, and 4,130 OTUs were identified in gut samples. Approximately half (51.84%) of the total OTUs were shared between the skin and gut microbiomes. A total of 397 OTUs were found to be unique to the skin communities, while 1,783 OTUs were found exclusively in the gut. After standardization, 405,090 skin sequences representing 2,441 OTUs, and 1,827,175 gut sequences representing 3,066 OTUs remained in the analysis. Regardless of sequence and OTU losses associated with standardization of samples, sequence coverage was ≥ 98% for all samples (Tables 1 and 2).
Table 1. Diversity indices of skin as calculated by Mothur (version 1.33.3). Operational taxonomic units (OTUs) were defined at 97% sequence similarity. Significance among total values for each fish species was determined by a one-way ANOVA followed by Tukey’s post hoc test. Within a column, different superscript letters mean significant difference (p < 0.05)
|
Species |
Sobs |
ACE |
Chao1 |
Shannon evenness |
Good’s coverage |
|
Bluegill |
496A |
1048 A |
812 A |
0.731 A |
0.983 |
|
Spotted gar |
455AB |
951 A |
754 A |
0.719 AB |
0.984 |
|
Largemouth bass |
390B |
930 A |
705 A |
0.660 B |
0.985 |
Table 2. Diversity indices of gut as calculated by Mothur (version 1.33.3). Operational taxonomic units (OTUs) were defined at 97% sequence similarity. Significance among total values for each fish species was determined by a one-way ANOVA followed by Tukey’s post hoc test. Within a column, different superscript letters mean significant difference (p < 0.05)
|
Group |
Sobs |
ACE |
Chao1 |
Shannon evenness |
Good’s coverage |
|
Bluegill |
705A |
1325A |
1101A |
0.525A |
0.994 |
|
Spotted Gar |
543BC |
1119AB |
912BC |
0.422BC |
0.995 |
|
Largemouth bass |
522C |
1044B |
840C |
0.449C |
0.996 |
Skin microbiome diversity. High sequence coverage for skin OTUs is reflected by the rarefaction curves generated by Mothur for each individual (Figure 1). Total expected richness as calculated by ACE and Chao1 showed no significant differences among species. However, the total observed OTUs and the Shannon evenness index (SEI) for bacterial communities of the skin were found to be significantly higher in Bluegill than in Largemouth Bass with Spotted Gar showing more variation in the number of observed OTUs (Table 1). Regarding sampling date, the number of observed OTUs, SEI, and the number of predicted OTUs as calculated by Chao1 were significantly higher for August than for November or May (data not shown). Rarefaction curves generated from each individual fish highlighted a larger alpha diversity in Spotted Gar than in blue gill and Largemouth Bass. In blue gill and Spotted Gar august skin communities seemed to transition from a higher diversity in August to a lesser diverse community in May and a transition phase in November. However, the diversity of the skin communities in Largemouth Bass appeared fairly stable with the exception of one individual in the month of November who had a significant more diverse community. Of the three sampling periods, May represented the least observed species richness in all species.
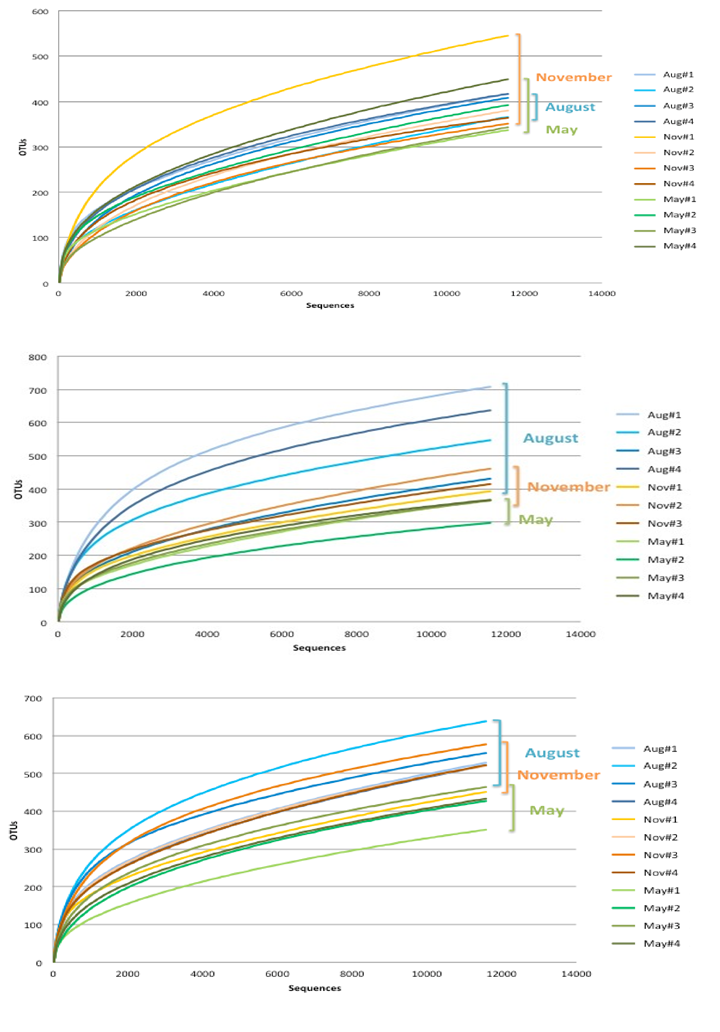
Figure 1. Rarefaction curves reflect species richness for the skin communities of each species. Panel A, Largemouth Bass; Panel B, Spotted Gar; Panel C, Bluegill. All sequences were standardized to the least number of obtained sequences for direct comparison. Curves are bracketed to show individual variations in richness by seasonal time points.
Skin microbiome composition. Overall, analysis of the sequence data revealed that the skin microbiomes for the three species consisted of 27 bacterial phyla with an additional 0.01% of unidentified phyla. Six phyla comprised the majority of sequences in all three species (Figure 2). Proteobacteria was the most common phylum in Blue Gill (40%), Spotted Gar (37%), and Largemouth Bass (35%). Following in abundance were the phyla Fusobacteria, Firmicute, Bacteroidetes, Deinococcus_thermus, Actinobacteria, and Tenericutes. Overall, more than 75% of the skin OTUs belonged to the phyla Proteobacteria, Fuseobacteria, Firmicute, or Bacteoidetes. Within the Proteobacteria, the most abundant class was Gammaproteobacteria (17%) followed by Betaproteobacteria (11%), Alphaproteobacteria (8%), Deltaproteobacteria (1%), and Epsilonproteobacteria (<0.1%). Twenty-one phyla (these counts not including representatives found in the “Spring Alpine Meadow” candidate division and other unidentified bacterial phyla) were present in varying abundances for all three species. The phylum Ignavibacteriae was only found in some of the skin samples from Bluegill while Dictyoglomi was only present in a single Spotted Gar sample.
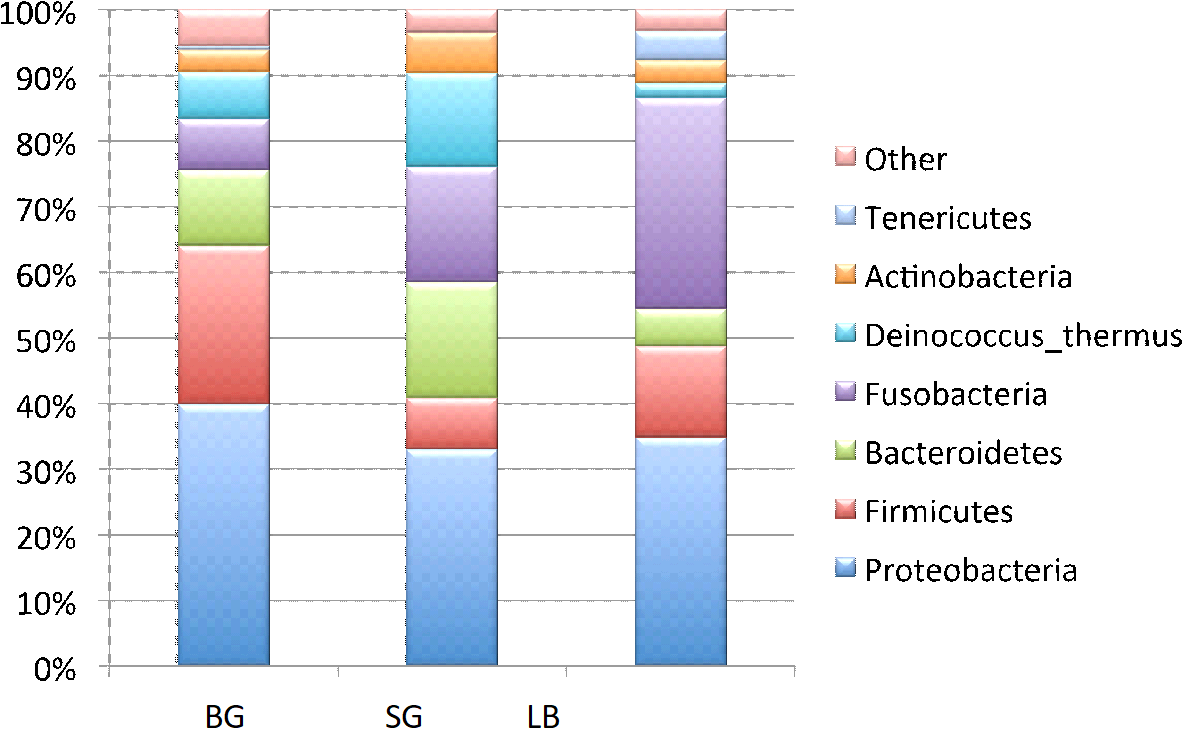
Figure 2. Overall percentage abundance of individual genera (Y axis) in the skin microbiome of Bluegill (BG), Spotted Gar (SG), and Largemouth Bass (LB).
Overall, 626 genera were found to populate the skin microbiomes for the three species. The top 10 genera included Cetobacterium (19.33%), Clostridium (10.39%), Deinococcus (9.36%), Plesiomonas (2.89%), Pseudomonas (2.89%), Cloacibacterium (2.51%), Bacteroides (2.32%), Aeromonas (2.06%), Vibrio (1.72%), and Acinetobacter (1.54%). The relative percent abundances in which these genera constituted the skin microbiomes varied between the species and across sampling points within each species (Table 3). Variations in abundances could also be seen between individuals both across sampling dates and within, but the specifics of this data are not reported herein.
Table 3. Top 10 genera identified from skin samples at each sampling time.
|
Bluegill - Overall |
|
Bluegill - August |
|
Bluegill - November |
|
Blue Gill - May |
||||
|
Genus |
% |
|
Genus |
% |
|
Genus |
% |
|
Genus |
% |
|
Clostridium |
17.64% |
Deinococcus |
17.03% |
Clostridium |
38.22% |
Cetobacterium |
17.11% |
|||
|
Cetobacterium |
7.71% |
Clostridium |
7.82% |
Turicibacter |
4.16% |
Bacteroides |
10.75% |
|||
|
Deinococcus |
7.07% |
Pseudomonas |
7.81% |
Pseudomonas |
3.87% |
Clostridium |
10.60% |
|||
|
Pseudomonas |
4.86% |
Cetobacterium |
5.39% |
Cyanobacterium |
2.78% |
Lysobacter |
6.81% |
|||
|
Bacteroides |
4.06% |
Cloacibacterium |
2.75% |
Acinetobacter |
2.55% |
Cloacibacterium |
6.53% |
|||
|
Cloacibacterium |
3.71% |
Janthinobacterium |
2.71% |
Cloacibacterium |
2.09% |
Thauera |
5.55% |
|||
|
Acinetobacter |
2.73% |
Exiguobacterium |
2.45% |
Shewanella |
1.78% |
Acinetobacter |
3.96% |
|||
|
Lysobacter |
2.48% |
Acinetobacter |
1.93% |
Microcystis |
1.55% |
Aeromonas |
3.94% |
|||
|
Aeromonas |
2.01% |
Sphingomonas |
1.92% |
Bacillus |
1.43% |
Acidovorax |
2.34% |
|||
|
Thauera |
1.97% |
Beijerinckia |
1.72% |
Janthinobacterium |
1.42% |
Hylemonella |
2.23% |
|||
|
Spotted Gar - Overall |
|
Spotted Gar - August |
|
Spotted Gar - November |
|
Spotted Gar - May |
||||
|
Genus |
% |
|
Genus |
% |
|
Genus |
% |
|
Genus |
% |
|
Cetobacterium |
19.76% |
Deinococcus |
25.84% |
Holospora |
7.63% |
Cetobacterium |
36.86% |
|||
|
Deinococcus |
15.93% |
Cetobacterium |
14.83% |
Oenococcus |
7.36% |
Plesiomonas |
5.03% |
|||
|
Clostridium |
4.24% |
Clostridium |
5.52% |
Vibrio |
6.04% |
Aeromonas |
4.06% |
|||
|
Plesiomonas |
2.87% |
Hymenobacter |
3.14% |
Deinococcus |
5.77% |
Cloacibacterium |
3.74% |
|||
|
Hymenobacter |
2.46% |
Knoellia |
3.01% |
Achromobacter |
5.46% |
Clostridium |
2.96% |
|||
|
Vibrio |
2.36% |
Sphingomonas |
2.53% |
Orientia |
4.63% |
Polynucleobacter |
2.90% |
|||
|
Sphingomonas |
2.02% |
Vibrio |
2.53% |
Hymenobacter |
4.19% |
Pseudomonas |
2.87% |
|||
|
Pseudomonas |
1.94% |
Plesiomonas |
2.34% |
Erythrobacter |
3.46% |
Hylemonella |
2.51% |
|||
|
Knoellia |
1.79% |
Bacteroides |
2.15% |
Cetobacterium |
2.69% |
Acinetobacter |
2.14% |
|||
|
Cloacibacterium |
1.59% |
Arthrobacter |
1.69% |
Candidatus odyssella |
2.41% |
Exiguobacterium |
2.11% |
|||
|
Largemouth Bass - Overall |
|
Largemouth Bass - August |
|
Largemouth Bass - November |
|
Largemouth Bass - May |
||||
|
Genus |
% |
|
Genus |
% |
|
Genus |
% |
|
Genus |
% |
|
Cetobacterium |
32.25% |
Cetobacterium |
36.24% |
Cetobacterium |
43.52% |
Cloacibacterium |
9.32% |
|||
|
Clostridium |
11.23% |
Clostridium |
7.77% |
Clostridium |
16.52% |
Aeromonas |
8.86% |
|||
|
Plesiomonas |
8.20% |
Vibrio |
6.73% |
Plesiomonas |
13.00% |
Hylemonella |
5.15% |
|||
|
Mycoplasma |
4.42% |
Plesiomonas |
5.75% |
Mycoplasma |
7.71% |
Acidovorax |
4.63% |
|||
|
Aeromonas |
3.06% |
Deinococcus |
5.29% |
Ferrovum |
2.01% |
Vogesella |
4.50% |
|||
|
Cloacibacterium |
2.52% |
Bacteroides |
4.53% |
Aeromonas |
2.00% |
Clostridium |
4.28% |
|||
|
Vibrio |
2.40% |
Pseudomonas |
2.50% |
Candidatus soleaferrea |
1.55% |
Dechloromonas |
3.91% |
|||
|
Deinococcus |
2.10% |
Mycoplasma |
1.74% |
Pseudomonas |
0.96% |
Pseudomonas |
3.77% |
|||
|
Pseudomonas |
2.04% |
Tetrasphaera |
1.25% |
Cyanobacterium |
0.70% |
Arthrobacter |
2.88% |
|||
|
Bacteroides |
1.62% |
|
Kocuria |
0.95% |
|
Cloacibacterium |
0.69% |
|
Acinetobacter |
2.87% |
Multidimensional scaling (MDS) plots based on skin OTU abundances were generated in effort to better visualize clustering patterns for each factor, fish species and sampling date. MDS plots indicated the bacterial composition of the skin was influenced more by sampling date than by fish species (Figure 3). These results were supported my ANOSIM for both fish species and sampling date (Table 4). Within each species, all groupings were well separated by date as confirmed by ANOSIM with global R values of 1.000, 0.817, and 0.867 for Bluegill, Largemouth Bass, and Spotted Gar, respectively. A two-way crossed ANOSIM (Table 4) was also run to detect possible interactions between fish species and sampling date. Overall analysis and pairwise tests indicate some type of interaction does exist between these two factors resulting in a higher level of separation between groups.
Table 4. One-way ANOSIM of the skin microbiome generated with Primer (v.6)
|
Pairwise tests |
R value |
p value |
|
By species |
|
|
|
Global |
0.179 |
0.002 |
|
Spotted gar vs. Largemouth bass |
0.081 |
0.110 |
|
Spotted gar vs. Bluegill |
0.326 |
0.001 |
|
Bluegill vs. largemouth bass |
0.142 |
0.036 |
|
By date |
|
|
|
Global |
0.583 |
0.001 |
|
August vs. November |
0.545 |
0.001 |
|
August vs. May |
0.609 |
0.001 |
|
November vs. May |
0.606 |
0.001 |
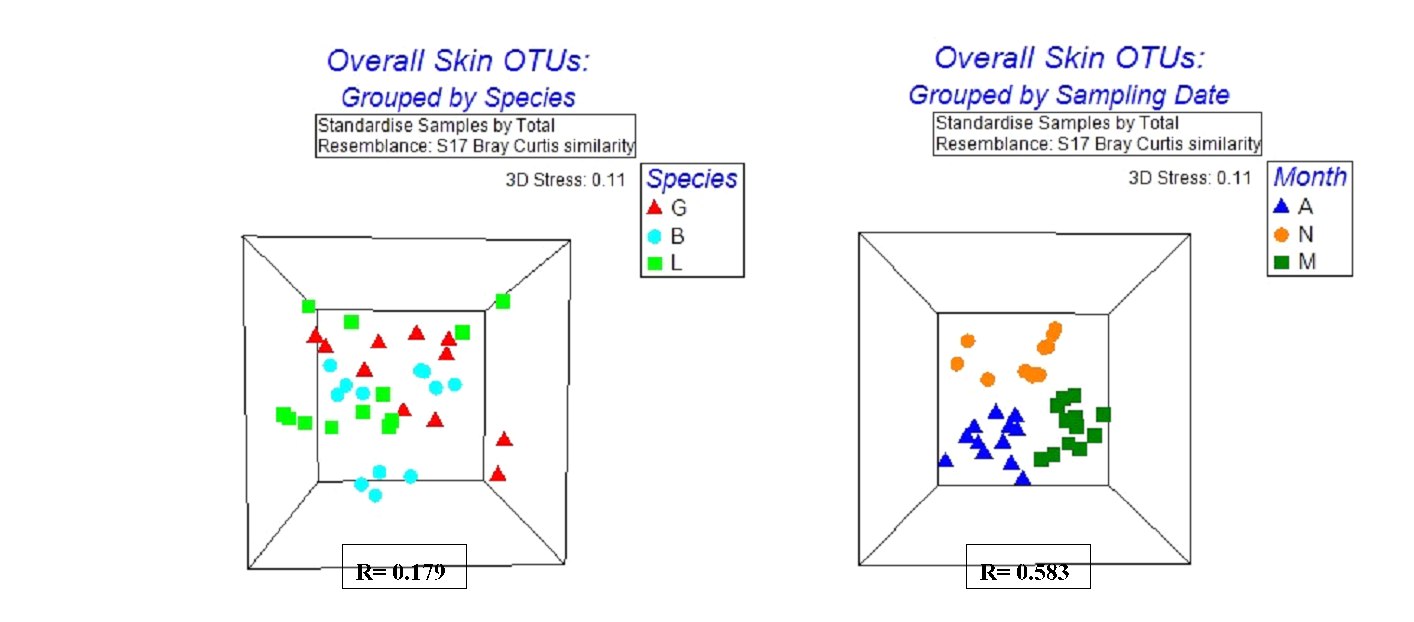
Figure 3. Multidimensional scaling (MDS) plots separated by factor for skin samples. ANOSIM results are indicated by R values for each factor. A, Fish species: Largemouth bass (L), Bluegill (B), and Spotted Gar (G); B, sampling date: August (A), November (N), and May (M).
Table 5 shows one-way crossed SIMPER analyses by bacterial genera responsible for the largest differences in the composition of skin communities between fish species. Relative percent abundances of Cetobacterium, Deinococcus, and Clostridium contributed to the highest percentages of dissimilarity for all pairwise groupings of fish species and sampling dates.
Table 5. One-way SIMPER analysis of the skin microbiome groups showing pairwise dissimilarity and main genera contributing to dissimilarity.
|
Average Abundance |
||||
|
Fish Species |
Bacteria Genus |
Species 1 |
Species 2 |
Contribution to dissimilarity (%) |
|
1. Bluegill |
Cetobacterium |
7.3 |
23.89 |
15.57 |
|
2. Largemouth Bass |
Clostridium |
16.49 |
9.27 |
8.66 |
|
Deinococcus |
6.01 |
2.93 |
4.62 |
|
|
Plesiomonas |
0.78 |
6.09 |
3.83 |
|
|
Cloacibacterium |
3.47 |
3.69 |
3.04 |
|
|
Bacteroides |
4.13 |
1.51 |
2.82 |
|
|
Acinetobacter |
4.04 |
1.35 |
2.66 |
|
|
Vibrio |
0.36 |
3.63 |
2.48 |
|
|
Mycoplasma |
0.69 |
3.54 |
2.47 |
|
|
Pseudomonas |
4.9 |
2.75 |
2.45 |
|
|
Ave. diss.= 70.99% |
Aeromonas |
2.07 |
3.75 |
2.39 |
|
1. Spotted Gar |
Cetobacterium |
19.21 |
23.89 |
17.45 |
|
2. Largemouth Bass |
Deinococcus |
11.42 |
2.93 |
7.55 |
|
Clostridium |
3.71 |
9.27 |
4.67 |
|
|
Plesiomonas |
2.98 |
6.09 |
3.97 |
|
|
Vibrio |
3.02 |
3.63 |
3.06 |
|
|
Cloacibacterium |
2.33 |
3.69 |
2.93 |
|
|
Aeromonas |
1.75 |
3.75 |
2.74 |
|
|
Ave. diss.= 70.43% |
Mycoplasma |
0.04 |
3.54 |
2.50 |
|
1.Spotted Gar |
Cetobacterium |
19.21 |
7.3 |
11.84 |
|
2. Bluegill |
Clostridium |
3.71 |
16.49 |
9.45 |
|
Deinococcus |
11.42 |
6.01 |
8.18 |
|
|
Bacteroides |
1.31 |
4.13 |
2.85 |
|
|
Acinetobacter |
1.13 |
4.04 |
2.52 |
|
|
Pseudomonas |
2.21 |
4.9 |
2.29 |
|
|
Cloacibacterium |
2.33 |
3.47 |
2.21 |
|
|
Ave. diss.= 69.96% |
Vibrio |
3.02 |
0.36 |
2.03 |
Gut Microbiome Diversity. Sequence coverage remained ≥ 99% for all samples (Table 2). High sequence coverage for skin OTUs was confirmed by rarefaction curves (data not shown). All diversity indices (i.e. observed OTUs, ACE, Chao1, and SEI) showed significant differences among the three species. Observed OTUs, SEI, and the predicted OTUs as calculated by Chao1 were significantly higher in Bluegill than in Spotted Gar and Largemouth Bass. Of the three species, Largemouth Bass again represented the lowest observed species richness (Table 2). Regarding sampling date, the total expected richness as calculated by ACE and Chao1 were significantly higher for August than for November or May. However, the observed richness and SEI showed no significant differences. Statistical analyses using two-way ANOVA indicated significant differences between the species and sampling period; however, no significant interaction was identified between these two variables and the gut microbiome (data not shown).
Gut microbiome composition. Similar to the skin microbiome, the overall gut microbiome was found to consist of 27 bacterial phyla with a minute percentage (0.0003%) of unidentified phyla. Not counting the incidence of unidentified bacterial taxa, 19 phyla were found to be present in the gut communities of all three species. Over 97% of the overall bacterial communities of the gut were composed primarily of four phyla: Fusobacteria (35.13%), Firmicutes (32.52%), Proteobacteria (15.87%), and Bacteroidetes (14.62%) (Figure 4). Each species of fish had at least one phylum present that was unique to its gut microbiome. Bluegill had the highest incidence of unique phyla present including the phyla Chlorobi (1 individual/month sampled), Elusimicrobia (1 individual in May), and Synergistetes (2 individuals in May). Other unique phyla found were Deferribacteres and Candidatus saccharibacteria (part of the “Spring Alpine Meadow” candidate division) identified in the gut communities of Spotted Gar and Largemouth Bass, respectively. Ignavibacteriae and Thermotogae were also found in very low abundances in a few gut samples from both the Bluegill and the Spotted Gar during November and May sampling dates.
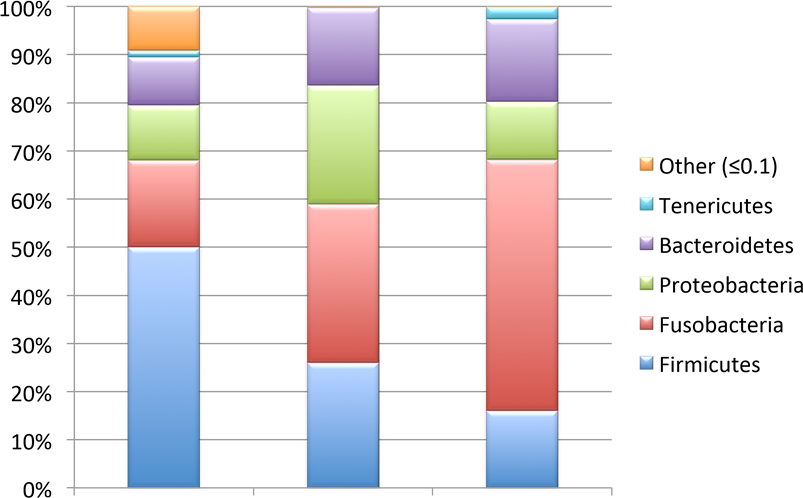
Figure 4. Overall percentage abundance of individual genera (Y axis) in the gut microbiome of Bluegill (BG), Spotted Gar (SG), and Largemouth Bass (LB).
In total, 804 genera were identified to inhabit the gut communities for the three species. The top 10 genera found in the gut microbiome for all samples were as follows: Cetobacterium (35.02%), Clostridium (26.97%), Bacteroides (13.66%), Plesiomonas (7.90%), Aeromonas (3.58%), Romboutsia (1.73%), Phyloobacterium (1.63%), Mycoplasma (1.42%), Turibacter (1.03%), and Ferrovum (1.00%). Although Cetobacterium was found to make up the highest percentages of the gut microbiome overall, the gut communities of Bluegill were dominated primarily by Clostridium (43.75%). Clostridium constituted over 51% of the gut microbiome during August and November; however, a shift was seen in May where Cetobacterium and Clostridium constituted 28.42% and 28.09% of bacterial communities of the gut, respectively. The relative percent abundances in which these genera constituted the gut microbiomes varied between the species and across sampling points within each species (Table 6). Variations in abundances could also be seen between individuals both across sampling dates and within, but the specifics of this data are not reported herein.
Table 6. Top ten genera identified from gut samples at each sampling point.
|
Bluegill - Overall |
|
Bluegill - August |
|
Bluegill - November |
|
Bluegill - May |
||||
|
Genus |
% |
|
Genus |
% |
|
Genus |
% |
|
Genus |
% |
|
Clostridium |
43.75% |
Clostridium |
53.76% |
Clostridium |
51.44% |
Cetobacterium |
28.42% |
|||
|
Cetobacterium |
19.63% |
Cetobacterium |
22.22% |
Aeromonas |
16.30% |
Clostridium |
28.09% |
|||
|
Bacteroides |
8.86% |
Turicibacter |
6.34% |
Cetobacterium |
7.56% |
Bacteroides |
17.13% |
|||
|
Aeromonas |
6.15% |
Romboutsia |
2.86% |
Bacteroides |
6.00% |
Romboutsia |
6.90% |
|||
|
Romboutsia |
3.95% |
Bacteroides |
2.30% |
Mycoplasma |
4.32% |
Plesiomonas |
5.06% |
|||
|
Plesiomonas |
2.58% |
Aeromonas |
1.65% |
Ferrovum |
3.54% |
Peptoclostridium |
4.21% |
|||
|
Turicibacter |
2.49% |
Ureibacillus |
1.59% |
Romboutsia |
1.74% |
Dysgonomonas |
2.18% |
|||
|
Peptoclostridium |
1.60% |
Candidatus rhabdochlamydia |
1.46% |
Bacillus |
1.58% |
Aeromonas |
0.77% |
|||
|
Ferrovum |
1.45% |
Limibacter |
1.30% |
Paludibacter |
1.41% |
Ferrovum |
0.70% |
|||
|
Mycoplasma |
1.44% |
Bacillus |
1.13% |
Plesiomonas |
1.32% |
Turicibacter |
0.67% |
|||
|
Spotted Gar - Overall |
|
Spotted Gar - August |
|
Spotted Gar - November |
|
Spotted Gar - May |
||||
|
Genus |
% |
|
Genus |
% |
|
Genus |
% |
|
Genus |
% |
|
Cetobacterium |
33.60% |
Cetobacterium |
48.79% |
Cetobacterium |
31.62% |
Clostridium |
29.20% |
|||
|
Clostridium |
21.89% |
Bacteroides |
22.89% |
Clostridium |
31.61% |
Cetobacterium |
15.69% |
|||
|
Bacteroides |
15.80% |
Plesiomonas |
13.13% |
Plesiomonas |
16.38% |
Plesiomonas |
15.30% |
|||
|
Plesiomonas |
14.58% |
Clostridium |
11.28% |
Ruminiclostridium |
6.24% |
Bacteroides |
13.02% |
|||
|
Phyllobacterium |
4.75% |
Aeromonas |
0.92% |
Bacteroides |
5.89% |
Phyllobacterium |
12.58% |
|||
|
Aeromonas |
2.27% |
Phyllobacterium |
0.48% |
Ferrovum |
2.20% |
Aeromonas |
4.32% |
|||
|
Ruminiclostridium |
1.44% |
Eubacterium |
0.46% |
Aeromonas |
1.70% |
Pantoea |
3.11% |
|||
|
Pantoea |
1.18% |
Cellulosilyticum |
0.31% |
Phyllobacterium |
0.83% |
Romboutsia |
1.55% |
|||
|
Romboutsia |
0.73% |
Ruminiclostridium |
0.22% |
Romboutsia |
0.58% |
Peptoclostridium |
1.09% |
|||
|
Ferrovum |
0.60% |
Romboutsia |
0.16% |
Bacillus |
0.38% |
Cellulosilyticum |
0.54% |
|||
|
Largemouth Bass - Overall |
|
Largemouth Bass - Aug |
|
Largemouth Bass - November |
|
Largemouth Bass - May |
||||
|
Genus |
% |
|
Genus |
% |
|
Genus |
% |
|
Genus |
% |
|
Cetobacterium |
51.80% |
Cetobacterium |
52.02% |
Cetobacterium |
59.41% |
Cetobacterium |
43.84% |
|||
|
Bacteroides |
16.71% |
Bacteroides |
33.02% |
Clostridium |
17.21% |
Clostridium |
20.37% |
|||
|
Clostridium |
14.30% |
Plesiomonas |
6.44% |
Mycoplasma |
6.69% |
Bacteroides |
14.70% |
|||
|
Plesiomonas |
7.58% |
Clostridium |
5.27% |
Aeromonas |
5.44% |
Plesiomonas |
13.49% |
|||
|
Mycoplasma |
2.60% |
Dysgonomonas |
0.40% |
Plesiomonas |
2.91% |
Phyllobacterium |
1.07% |
|||
|
Aeromonas |
2.09% |
Asaccharospora |
0.38% |
Bacteroides |
2.64% |
Mycoplasma |
0.85% |
|||
|
Ferrovum |
0.87% |
Peptoclostridium |
0.24% |
Ferrovum |
2.29% |
Turicibacter |
0.84% |
|||
|
Phyllobacterium |
0.56% |
Mycoplasma |
0.18% |
Fusobacterium |
0.65% |
Romboutsia |
0.66% |
|||
|
Romboutsia |
0.35% |
Vogesella |
0.15% |
Phyllobacterium |
0.59% |
Aeromonas |
0.63% |
|||
|
Turicibacter |
0.34% |
|
Aeromonas |
0.15% |
|
Candidatus soleaferrea |
0.30% |
|
Dysgonomonas |
0.42% |
Multidimensional scaling (MDS) plots based on gut OTU abundances were generated in an effort to better visualize clustering patterns for the designated factors, fish species and sampling date. Results indicated the bacterial composition of the gut was again influenced more by sampling date than by fish species (Figure 5). Within each species, Bluegill and Largemouth Bass showed moderate to high separation by sampling date as supported by ANOSIM with global R values of 0.690 (separated but overlapping groups) and 0.813 (well separated), respectively. However, the Spotted Gar had a lot more variability between replicates and showed no significant differences between sampling dates (R=0.187. A two-way crossed ANOSIM (data not shown) was also run to pick up on possible interactions between the fish species and the sampling date. Overall analysis and pairwise tests indicate some type of interaction does exist between these two factors resulting in a higher level of separation between groups.
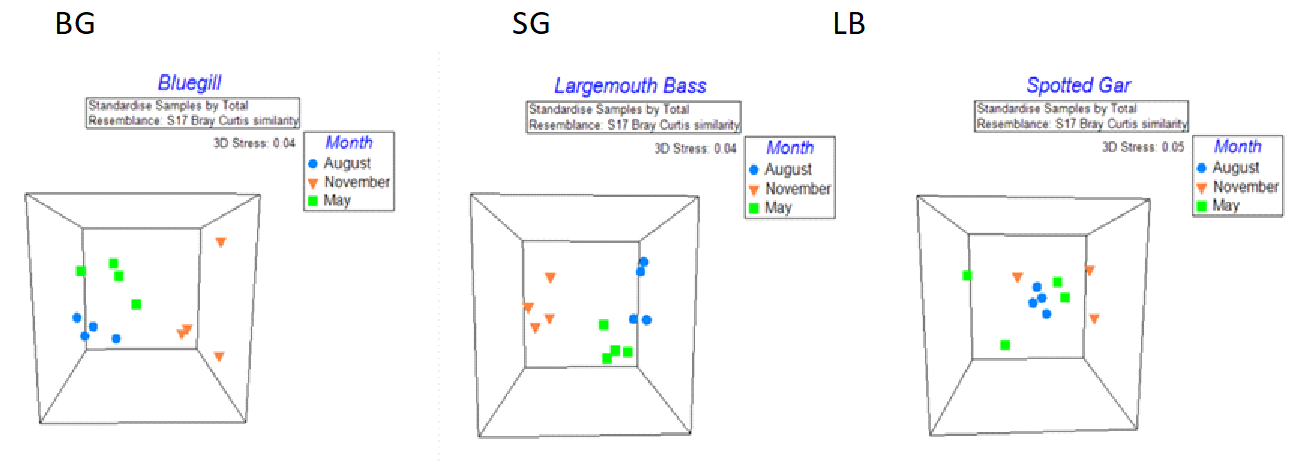
Figure 5. Multidimensional scaling (MDS) plots for each fish species demonstrating grouping of individual gut samples by sampling date.
Similar to analyses for the skin microbiome, one-way crossed SIMPER analyses by bacterial genera found the largest differences in the composition of gut communities between fish species for Bluegill and Largemouth Bass as well as sampling dates for November and May. The bacterial genera Cetobacterium, Clostridium, Bacteroides, and Plesiomonas accounted for the highest percentages of dissimilarity for all pairwise groupings between fish species (Table 7).
Table 7. SIMPER analysis comparing gut communities by sampling date. Only genera accounting for at least 2% of dissimilarity between each combination of sampling month is given including average abundances for each month sampled as well as the percent contribution to dissimilarity for each genus.
|
One-way SIMPER Analysis of the Gut Microbiome: Genus level by Sampling Date |
||||
|
Average Abundance |
||||
|
Fish Species |
Bacteria Genus |
Month 1 |
Month 2 |
Contribution to dissimilarity (%) |
|
1. November |
Cetobacterium |
34.04 |
27.70 |
23.49 |
|
2. May |
Clostridium |
33.72 |
26.39 |
21.95 |
|
Bacteroides |
4.67 |
13.80 |
11.57 |
|
|
Plesiomonas |
5.09 |
12.78 |
11.31 |
|
|
Aeromonas |
7.75 |
1.56 |
6.61 |
|
|
Phyllobacterium |
0.59 |
5.12 |
4.88 |
|
|
Mycoplasma |
4.13 |
0.33 |
3.71 |
|
|
Romboutsia |
0.93 |
3.37 |
3.09 |
|
|
Ave. diss.= 55.40% |
Ferrovum |
2.72 |
0.46 |
2.18 |
|
1. August |
Clostridium |
22.98 |
33.72 |
26.23 |
|
2. November |
Cetobacterium |
41.88 |
34.04 |
24.46 |
|
Bacteroides |
19.39 |
4.67 |
16.22 |
|
|
Plesiomonas |
6.41 |
5.09 |
7.25 |
|
|
Aeromonas |
0.91 |
7.75 |
6.93 |
|
|
Mycoplasma |
0.09 |
4.13 |
3.87 |
|
|
Ferrovum |
0.07 |
2.72 |
2.50 |
|
|
Ave. diss.= 53.01% |
Turicibacter |
2.14 |
0.40 |
2.07 |
|
1. August |
Clostridium |
22.98 |
26.39 |
24.15 |
|
2. May |
Cetobacterium |
41.88 |
27.7 |
23.82 |
|
Bacteroides |
19.39 |
13.8 |
16.57 |
|
|
Plesiomonas |
6.41 |
12.78 |
11.00 |
|
|
Phyllobacterium |
0.16 |
5.12 |
5.19 |
|
|
Romboutsia |
1.05 |
3.37 |
3.62 |
|
|
Ave. diss.= 49.89% |
Turicibacter |
2.14 |
0.50 |
2.23 |
In recent years, a paradigm shift has occurred in the area of organismal health and pathogenesis (Vayssier-Taussat et al., 2014). Where Koch and Hill’s fundamental postulates equating to “one microbe—one disease” were once regarded as the rule, research has now shifted to a more holistic view in which whole microbial communities give rise to and participate in complex interactions that can ultimately impact and fuel disease processes. Due largely to the ever-growing body of research concerning mammalian species, the bacterial communities that comprise the microbiomes of various internal and external surfaces of the body are now recognized as integral components to the overall health of the host. Historically somewhat hampered by culture-dependent methods that often produced incomplete or biased results, research regarding microbiomes is now conceivably one of the fastest developing fields in biology. With the advent of newer, more advanced sequencing technologies and data processing platforms, researchers are now able to more fully unearth the cryptic diversity and function of these microbial communities much more quickly and at a fraction of the cost (Llewellyn et al., 2014; Tarnecki et al., 2017). Although research lags far behind mammalian microbiome studies, the bacterial communities that constitute fish microbiomes are now considered to be essential components in host health, nutrition, growth and development, and defense against invading pathogens (Austin, 2006; Nayak, 2010).
The composition at the phylum level of the bacterial communities found in association with fish skin that we described in this study was consistent with previous findings from skin and mucus of various fish taxa including freshwater and saltwater environments (Cipriano and Dove, 2009; Arias et al., 2013; Larsen et al., 2013; Larsen et al., 2015; Mohammed and Arias, 2015). However, at the genus level, there were some surprising results, as Cetobacterium (19.33%) was found to be the most abundant genus overall for skin communities of the three species (Table 4). Within each species, the highest abundance of this genus was seen in M. salmoides (32.25%), with Cetobacterium representing the highest percentages of bacterial sequences for August (36.24%) and November (43.52%). Finding this bacterial genus in such appreciable numbers as part of the skin microbiome is surprising since the vast majority of reports for this genus have typically been associated with the gastrointestinal tracts of mammals and other fishes (Tsuchiya et al., 2008; Larsen et al., 2014a; Li et al., 2014). In the gut environment, some strains of Cetobacterium are capable of producing high amounts of vitamin B12 and also inhibiting growth of some other bacterial taxa. Cetobacterium is microaerotolerant (although some strain can grow in up to 6% oxygen) (Finegold et al., 2003) thus fish skin is unlikely to be a natural habitat for this genus. Although most reports of this genus have been associated with the gastrointestinal tracts of various organisms, Cetobacterium has also been reported in small percentages (2.19% of sequence abundance) in earthen pond water samples from an aquaculture facility in China (Li et al., 2014).
For purposes of this study, Lake Guntersville was considered to be a single hydrological unit. In this respect, all collection sites were compared equally over the course of the study based on the idea that fishes in their dynamic, natural habitats are not confined to static locations. From this perspective, seasonality was shown to significantly influence both the skin and gut microbiome structures of fishes in Lake Guntersville. Sampling date resulted in a higher degree of separation between samples than the species level in skin communities (R=0.583) and to a lesser extent in the gut communities (R=0.391) indicating these communities may be more stable than in the skin. Our group has shown a marked specificity between the bacterial communities associated with skin and their hosts; however, environmental factors such as water temperature and salinity definitely exert a stronger effect on the bacterial communities exposed to the elements than to those protected by the more stable gut environment.
Micropterus salmoides was shown to have the least bacterial diversity for both the skin and gut microbiomes when compared to L. macrochirus and L. oculatus, with significant differences being found in the diversity of L. macrochirus and M. salmoides in both cases. Larsen et al. (2014a) found similar results in an experimental recreational fishing pond from which fecal contents of channel catfish (Ictalurus punctatus), bluegill, and largemouth bass were sequenced via pyrosequencing techniques and compared. In both cases, Bluegill was observed to have significantly higher observed OTUS, expected richness, and evenness when compared to Largemouth Bass. While the specific stomach and fecal contents of the fish species used in this study were not analyzed, Bluegill are generalists that typically had greater incidences of plant materials in fecal samples, while the contents of the Largemouth Bass (piscivorous) were typically more consistent with digested vertebrate species. In mammalian studies, both the host phylogeny and the diet have been implicated as potential factors influencing the bacterial diversity of gut microbiomes, with herbivores typically demonstrating higher bacterial diversity than carnivores (Ley et al., 2008). Also worth mentioning, similar to Larsen et al. (2014a), Largemouth Bass were shown to have high abundances of Cetobacterium in the gut, averaging close to 52% composition over the course of the study. The gut microbiomes of Bluegill from this reservoir, on the other hand, demonstrated a higher proportion of Clostridium than what has been previously reported (Larsen et al., 2014a). These findings are consistent with a study performed by Liu et al. (Liu et al., 2016) where cellulose-degrading bacteria such as Clostridium were found to dominate the gut communities of herbivorous fishes, while carnivores were dominated more often by Cetobacterium as well as other protease-producing bacteria.
To the best of our knowledge, this research provides the first characterization of the skin and gut microbiomes of Spotted Gar. Largemouth Bass and Bluegill were selected largely for their high commercial value in terms of ecotourism (i.e. competitive and recreational fisheries) as well as for use as food fishes. Recently, sequencing of the highly conserved Spotted Gar genome has been shown to provide an important link between human biology and teleost biomedical models, illuminating the evolution of immunity, mineralization, and development (Braasch et al., 2016). By creating large-scale meiotic maps, Amores et al. (2011) confirmed that the spotted gar lineage diverged from teleosts prior to the teleost genome duplication (TGD). This research indicated that, although biologically more similar to teleosts, the organization of the spotted gar genome was actually somewhat more comparable to humans than teleosts. Subsequently, the study named the spotted gar as “the species of choice” for genomics, physiology, and development studies due largely to its phylogenetic position, accessibility and abundance, feasibility of spawning and rearing to adulthood under experimental conditions, and its slowly evolving genome.
Recent studies on evolutionary biology suggest that hosts and their microbiomes should be considered a single ecological unit – the holobiont (Richardson, 2017). Zilber-Rosenberg and Rosenberg (Zilber-Rosenberg and Rosenberg, 2008) defined the hologenome theory of evolution which views the holobiont as a mode of selection in evolution. From this perspective, the microbiome of a species is essentially viewed as an extension of the host genome, with host-microbe interactions being strongly implicated in the fundamental processes of adaptation and speciation (Zilber-Rosenberg and Rosenberg, 2008; Brucker and Bordenstein, 2011). This is particularly relevant since the microbiome can respond in more ways and more quickly than the genome of a host to environmental dynamics. Given the significant insights the spotted gar provides regarding the development and evolution of vertebrates from a genetics perspective, perhaps future research of this species may also have the potential to provide important clues into the mechanisms that have allowed for the colonization and coevolution of microbial symbionts on or within a host.
The overall goal for this study was to perform an in-depth characterization of the autochthonous microbial communities associated with (apparently) healthy Largemouth Bass, Bluegill, and the Spotted Gar. Despite the economic importance of these species, particularly Largemouth Bass, little information is available about their natural symbionts. We have provided baseline data for the type and abundance of bacteria that can be found associated with these three species. More comprehensive studies including spatiotemporal relationships between environmental conditions and fish microbiomes are required to further characterize those bacterial communities and, ultimately, identify dysbiosis (microbial imbalance or impaired microbiome negatively impacting host health) caused by anthropomorphic insults.
We would like to thank Dr. Matt Catalano, School of Fisheries, Auburn University, and his group for helping us collecting the animals. This work was partially funded by the Southeastern Cooperative Fish Parasite and Disease Project. In addition, the Alabama Experiment Station through USDA-HATCH funds supported this study.
Amores, A., Catchen, J., Ferrara, A., Fontenot, Q. & Postlethwait, J. H. (2011). Genome evolution and meiotic maps by massively parallel DNA sequencing: spotted gar, an outgroup for the teleost genome duplication. Genetics 188, 799-808. PMid:21828280 PMCid:PMC3176089
View Article PubMed/NCBIArias, C. R., Koenders, K. & Larsen, A. M. (2013). Predominant bacteria associated with red snapper Lutjanus campechanus (Poey, 1860) from the northern Gulf of Mexico. Journal of Aquatic Animal Health 25, 281-289. PMid:24341770
View Article PubMed/NCBIAustin, B. (2006). The bacterial microflora in fish, revised. TheScientificWorld Journal 6, 931-945. PMid:16906326
View Article PubMed/NCBIBackhed, F., Ley, R. E., Sonnenburg, J. L., Peterson, D. A. & Gordon, J. I. (2005). Host-bacterial mutualism in the human intestine. Science 307, 1915-1920. PMid:15790844
View Article PubMed/NCBIBalcazar, J. L., de Blas, I., Ruiz-Zarzuela, I., Cunningham, D., Vendrell, D. & Muzquiz, J. L. (2006). The role of probiotics in aquaculture. Veterinary Microbiology 114, 173-186. PMid:16490324
View Article PubMed/NCBIBraasch, I., Gehrke, A. & Postlethwait, J. H. (2016). The spotted gar genome illuminates vertebrate evolution and facilitates human-teleost comparisons. Nature Genetics 48, 427-437. PMid:26950095
View Article PubMed/NCBIBrucker, R. M. & Bordenstein, S. T. (2011). The roles of host evolutionary relationships (genus: Nasonia) and development in structuring microbial communities. Evolution 66, 349-362. PMid:22276533
View Article PubMed/NCBIChiarello, M., Villeger, S., Bouvier, C., Bettarel, Y. & Bouvier, T. (2015). High diversity of skin-associated bacterial communities of marine fishes is promoted by their high variability among body parts, individuals and species. FEMS Microbial Ecology 91, fiv061. PMid:26048284
View Article PubMed/NCBICipriano, R. C. & Dove, A. (2009). Far from superficial: microbial diversity associated with the dermal mucus of fish. 3rd Bilateral Conference.
Clements, K. D., Pasch, I. B. Y., Moran, D. & Turner, S. J. (2007). Clostridia dominate 16S rRNA gene libraries prepared from the hindgut of temperate marine herbivorous fishes. Marine Biology 150, 1431-1440.
View ArticleDeSantis, T., Hugenholtz, P., Larsen, N., Rojas, M., Brodie, E., Keller, K., Huber, T., Dalevi, D., Hu, P. & Andersen, G. G., a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Applied and environmental microbiology 72: 5069-5072 (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Applied and environmental microbiology 72, 5069-5072. PMid:16820507
View Article PubMed/NCBIDhanasiri, A. K., Bruvold, L., Brinchmann, M. F., Korsnes, K., Bergh, O. & Kiron, V. (2010). Changes in the instestinal microbiota of wild Atlantic cod Gadus morhua L. upon captive rearing. Microbial Ecology 61, 20-30. PMid:20424834
View Article PubMed/NCBIEllis, A. E. (1999). Immunity to bacteria in fish. Fish & Shellfish Immunology 9, 291-308.
View ArticleEsteban, M. A. (2012). An overview of the immunological defenses in fish skin. ISRN Immunology 2012, ID 853470
Finegold, S. M., Vaisanen, M.-L., Molitoris, D. R., Tomzynski, T. J., Song, Y. L., Liu, C., Collins, M. D. & Lawson, P. A. (2003). Cetobacterium somerae sp. nov. from human feces and emended description of the genus Cetobacterium. Systematic and Applied Microbiology 26, 177-181. PMid:12866843
View Article PubMed/NCBIHird, S. (2017). Evolutionary biology needs wild microbiomes. Frontiers in Microbiology 8, 725. PMid:28487687
View Article PubMed/NCBIHooper, L. V. (2009). Do symbiotic bacteria subvert host immunity? Nature Review 7, 367-374.
View ArticleHorsley, R. W. (1977). A review on the bacterial flora of teleosts and elamobrachs, includings methods for its analysis. Journal of Fish Biology 10, 529-553.
View ArticleHuber, I., Spanggaard, B., Appel, K. F., Rossen, L., Nielsen, T. & Gram, L. (2004). Phylogenetic analysis and in situ identification of the intestinal microbial community of rainbow trout (Oncorhynchus mykiss, Walbaum). Journal of Applied Microbiology 96, 117-132. PMid:14678165
View Article PubMed/NCBILarsen, A. M., Bullard, S. A., Womble, M. & Arias, C. R. (2015). Community structure of skin microbiome of Gulf killifish, Fundulus grandis, is driven by seasonality and not exposure to oiled sediments in a Lousiana salt marsh. Microbial Ecology 70, 534-544. PMid:25704317
View Article PubMed/NCBILarsen, A. M., Mohammed, H. H. & Arias, C. R. (2014a). Characterization of the gut microbiota of three commercially valuable warmwater species. Journal of Applied Microbiology 116, 1396-1404. PMid:24529218
View Article PubMed/NCBILarsen, A. M., Mohammed, H. H. & Arias, C. R. (2014b). Comparison of DNA extraction protocols for the analysis of gut microbiota in fishes. Fems Microbiology Letters 362, fnu031. PMid:25757730
PubMed/NCBILarsen, A. M., Tao, Z., Bullard, S. A. & Arias, C. R. (2013). Diversity of the skin microbiotas of fishes: evidence for host species specificity. FEMS Microbiology Ecology in press. PMid:23607777
View Article PubMed/NCBILederberg, J. (2001). 'Ome sweet' omics - A genealogical treasury of words. Scientist 15, 7.
Ley, R. E., Hamady, M., Lozupone, C. A., Turnbaugh, P., Ramey, R. R., Bircher, J. S., Schlegel, M. L., Tucker, T. A., Schrenzel, M. D., Knight, R. & Gordon, J. I. (2008). Evolution of mammals and their gut microbiomes. Science 320, 1647-1651. PMid:18497261 PMCid:PMC2649005
View Article PubMed/NCBILi, J., Ni, J., Li, J., Wang, C., Li, X., Wu, S., Zhang, T., Yu, Y. & Yan, Q. (2014). Comparative study of gastrointestinal microbiota of eight fish species with different feeding habits. Journal of Applied Microbiology 117, 1750-1760. PMid:25294734
View Article PubMed/NCBILittman, D. R. & Pamer, E. G. (2011). Role of commensal microbiota in normal and pathogenic host immune responses. Cell Host & Microbe 10, 311-323. PMid:22018232
View Article PubMed/NCBILiu, H., Guo, X., Gooneratne, R., Lai, R., Zeng, C., Zhan, F. & Wang, W. (2016). The gut microbiome and degradation enzyme activity of wild freshwater fishes influenced by their trophic levels. Scientific Reports 6, 24340.
View ArticleLlewellyn, M., Boutin, S., Hossein-Hoseinifar, S. & Derome, N. (2014). Teleost microbiomes: the state of the art in their characterization, manipulation and importance in aquaculture and fisheries. Frontiers in Microbiology 5, 1-17. PMid:24917852
View Article PubMed/NCBILockesh, J. & Kiron, V. (2016). Transition from freshwater to seawater reshapes the skin-associated microbiota of Atlantic salmon. Scientific reports 6, 19707. PMid:26806545
View Article PubMed/NCBIMohammed, H. H. & Arias, C. R. (2015). Potassium permanganate elicits a shift of the external microbiome and increases host susceptibility to columnaris disease. Veterinary Research 46, 82. PMid:26170019
View Article PubMed/NCBIMouchet, M. A., Bouvier, C., Bouvier, T., Troussellier, M., Escalas, A. & Mouillot, D. (2011). Genetic difference but functional similarity among fish gut bacterial communities through molecular and biochemical fingerprints. FEMS Microbiology Ecology 79, 568-580. PMid:22092438
View Article PubMed/NCBINayak, S. K. (2010). Role of gastrointestinal microbiota in fish. Aquaculture Research 41, 1553-1573
View ArticlePeatman, E., Lange, M., Zhao, H. & Beck, B. H. (2015). Physiology and immunology of mucosal barriers in catfish (Ictalurus spp.). Tissue Barriers, e1068907-1068901.
View ArticleRhee, K. J., Sethupathi, P., Driks, A., Lanning, D. K. & Knight, K. L. (2004). Role of commensal bacteria in development of gut-associated lymphoid tissues and preimmune antibody respertoire. The Journal of Immunology 172, 1118-1124. PMid:14707086
View Article PubMed/NCBIRichardson, L. A. (2017). Evolving as a holobiont. PLoS One 15, e2002168.
View ArticleRosello-Mora, R. (2005). Updating prokaryotic taxonomy. Journal of Bacteriology 187, 6225-6257.
View ArticleSchloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., Lesniewski, R. A., Oakley, B. B., Parks, D. H., Robinson, C. J., Sahl, J. W., Stres, B., Thallinger, G. G., Van Horn, D. J. & Weber, C. F. (2009). Introducing Mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and Environmental Microbiology 75, 7537-7541. PMid:19801464
View Article PubMed/NCBITarnecki, A. M., Burgos, F. A. & Arias, C. R. (2017). Fish intestinal microbiome: diversity and symbiosis unravelled by metagenomics. Journal of Applied Microbiology, doi: 10.1111/jam.13415.
View ArticleTsuchiya, C., Sakata, T. & Sugita, H. (2008). Novel ecological niche of Cetobacterium somerae, an anaerobic bacterium in the intestinal tracts of freshwater fish. Letters in Applied Microbiology 46, 43-48. PMid:17944860
PubMed/NCBITurnbaugh, P. J., Ley, R. E., Fraser-Liggett, C. M., Knight, R. & Gordon, J. I. (2007). The human microbiome project. Nature 449, 804-810. PMid:17943116
View Article PubMed/NCBIUchii, K., Matsui, K., Yonekura, R., Tani, K., Kenzaba, T., Nasu, M. & Kawabata, Z. (2006). Genetic and physiological characterization of the intestinal bacterial microbiota of bluegill (Lepomis macrochirus) with thre different feeding habits. Microbial Ecology 51, 277-283. PMid:16596440
View Article PubMed/NCBIVayssier-Taussat, M., Albina, E., Citti, C., Cosson, J. F., Jacques, M.-A., Lebrun, M.-H., Ogliastro, M., Petit, M.-A., Roumagnac, P. & Candresse, T. (2014). Shifting the paradigm from pathogens to pathobiome: new concepts in the light of meta-omics. Frontiers in Cellular and Infection Microbiology 4, 29. PMid:24634890
View Article PubMed/NCBIWells, C. L., Maddaus, M. A., Jechorek, R. P. & Simmons, R. L. (1988). Role of intestinal anaerobic bacteria in colonization resistance. European Journal of Clinical Microbiology and Infectious Diseases 7, 107-113.
View ArticleZilber-Rosenberg, I. & Rosenberg, E. (2008). Role of microorganisms in the evolution of animals and plants: the hologenome theory of evolution. FEMS Microbiology Reviews 32, 723-735. PMid:18549407
View Article PubMed/NCBI