Paula C. Castilho
E-mail: castilho@uma.pt
© 2019 Sift Desk Journals. All Rights Reserved
VOLUME: 1 ISSUE: 2
Page No: 34-37
Paula C. Castilho
E-mail: castilho@uma.pt
João C. Serina*, Paula C. Castilho*1, Miguel X. Fernandes*
*Centro de Química da Madeira, Universidade da Madeira, Campus da Penteada, 9020-105 Funchal (Portugal)1
Jos
Caffeoylquinic acids are ubiquitous phenolic compounds with several health benefits to humans and they have been shown to be promisinganti-HIV compounds. In this work, molecular docking was used to study the inhibition of HIV-I integrase and protease using caffeoylquinic acids. It was possible to establish that the naturally occurring caffeoylquinic acids are not suitable as inhibitors for protease but are very good inhibitors for integrase. A new binding site was found for 3, 4-O-di-Caffeoylquinic acid between the chains of HIV-I integrase that could possibly lead to a disruption of the catalytic process of HIV-I integrase.
KEYWORDS: HIV, integrase, protease, molecular docking, in silico, caffeoylquinic acid, enzyme inhibition
Caffeoylquinic acids (CQAs) are esters of caffeic and quinic acid. These hydroxycinnamates have drawn a lot of research attention in applications related to diabetes(1,2), cancer(3,4) and cardio-vascular diseases(5,6). In the 80s, CQAs were considered a novel family of anti-viral compounds(7) and much research was performed on their anti-HIV properties during the 90s(8–10). Recently, the interest in the mechanism of action of these substances over HIV has reappeared (11).
HIV-I protease (EC 3.4.23.16) is a key therapeutic target against HIV-I and, since its structure was discovered, FDA has approved ten inhibitors(12). Most of the inhibitors for proteases are modified peptides or peptide-like. Tipranavir® is the only FDA approved protease inhibitor that does not have a peptide scaffold. In addition to the low oral bioavailability and fast excretion, these inhibitors are becoming less effective with the emergence of drug resistant HIV (13). The design of new inhibitors is moving from these towards macro-cyclic molecules that retain the geometry of a β-strand (protease’s substrate) but without the peptide nature (13,14) in the hope that these new drugs will be able to overcome the many drug resistance mutations of HIV-I (15).
HIV-I integrase ensures that the genetic material of HIV is inserted into the genetic material of the host. Inhibiting it would prevent this from happening. In 1999,Zhu et al.(16) established that dicaffeoylquinic acids (diCQAs) could inhibit HIV-I integrase in an irreversible manner. They were also able to demonstrate that diCQAs were noncompetitive inhibitors of HIV-I integrase and several other Integrase enzymes. In 2010,Hu et al.(17) reported that 3, 4-O-diCQA had two possible binding mechanisms on HIV-I integrase. One of the binding sites was the active site of HIV-I integrase and the other binding site was in a pocket on the other side of the flexible loop that contains the catalytic residues. According to Hu et al., by binding at this site, 3, 4-O-diCQA would change the conformation of the loop and the enzyme was inhibited.
To perform the experimental work, the crystal structures of HIV-I protease and integrase were downloaded from the PDB website (ids: 1HXB and 3AO3 respectively).
The published crystal structure for protease refers to the enzyme containing the inhibitor Saquinavir and the crystal structure for the integrase contains a novel inhibitor referred to as 3-(1,3-benzodioxol-5-yl)-1-methyl-1H-pyrazole- 5-carboxylic acid. The conformations of each inhibitor were reproduced using AutoDockVina(18) and their binding affinity was used as control/base value to compare with the CQAs.
Molecules of severaldiCQAs(1,3-, 1,5-, 3,4-, 3,5- and 4,5-diCQA) were created and optimized using the MM+ force field to an RMS below 0.01kcal/mol. The molecule of 5-O-CQA was also used.
Three types of docking were used on this study. In rigid docking, the possible binding conformations of a ligand (small molecule) in a specific area of the receptor are evaluated by rotating its bonds and translating the molecule. Flexible docking is similar to rigid docking in the sense that the search area is specific but on this type of docking, several amino acid residues possess rotational freedom (several bonds can rotate). The rotation of the amino acids allows to reproduce more closely the accommodation of ligands when they bind to their target and also makes it possible to optimize results from rigid docking. Finally, blind docking has no flexibility on the amino acid residues but the search area includes the entire receptor. This type of docking is used to find the most likely place for a ligand to bind to the receptor, it is very useful to confirm results from other dockings or to find the binding site of a new ligand without the location bias of the rigid docking.
All docking calculations were performed using AutoDockVina(18).
The docking studies for HIV-I protease show that diCQAs will preferably bind to the same site as the commercial inhibitor Saquinavir®(fig 1 and 2) but with an affinity that was only 50-60% of the commercial product (fig 3).
Out of the analyzed CQAs, 1,3- and 1,5-diCQA had very similar conformations (fig 2 a). In addition, the 5’ branch of 3,5-diCQA and 5-CQA also had similar conformations, differing only on the 3-caffeoyl moiety position (fig 2 c). Both the rigid and the blind docking results show that the CQA that binds better to HIV-I protease (i. e. has the most negative affinity) is 3,5-diCQA as it is shown by the graph on fig 3. This indicates that, out of all the analyzed ligands, it is the one most likely to bind to HIV-I protease.
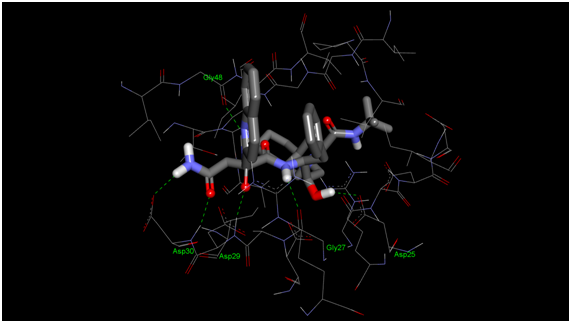
Fig. 1 - Saquinavir binding site on HIV-I protease (from pdb structure 1HXB)
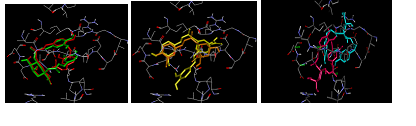
Fig. 2 a, b and c: Binding of CQAs to HIV-I protease, a)a:1,3-diCQA (red) and1,5-diCQA (green), b) 3,5-diCQA (yellow) and , c) 4,5-diCQA (magenta) and 3,4-diCQA (cyan)
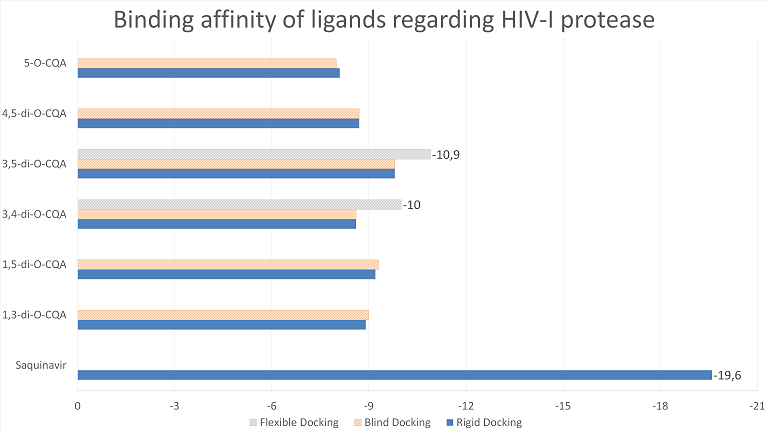
Fig.3 Binding affinity of ligands regarding HIV-I Protease (kcal/mol) – more negative values are better.
The crystal structure of HIV-I integrase had two sites containing the same inhibitor (3-(1,3-benzodioxol-5-yl)-1-methyl-1H-pyrazole- 5-carboxylic acid –one on chain A and one on B). This inhibitor was used as a control to compare with the affinity of the CQAs. Despite the fact that both chains of the enzyme were symmetrical, all ligands had better binding affinities on the B chain site than on the A chain. However, the control ligand had a lower binding affinity on the B chain (fig. 4). Regarding the best scoring conformation of the blind docking, 1,3-, 3,5- and 4,5-diCQAs were docked at the B chain pocket, 1,5-diCQA was docked at the A chain pocket and both 3,4-diCQA and 5-CQA were docked in a new site (fig. 5).
As it is possible to see on figure 5, while the inhibitor from the crystal structure of HIV-I integrase binds to pockets between both protein chains. The new binding site of 3,4-diCQA and 5-CQA is located at the top of the enzyme between both chains. In order to better visualize the binding of 3, 4-diCQA, it is represented by a surface wireframe. The residues that allow 3, 4-diCQA to bind to this novel site of HIV-I integrase through Hydrogen bonds are: A-Lys-71, A-Lys103, B-Lys-103, A-Lys-173 and B-Lys-173. As for 5-CQA, it established Hydrogen bonds with: A-Tyr-99, A-Lys-103, B-Lys-171, B-Lys-172, A-Lys-173 and B-Lys-173.
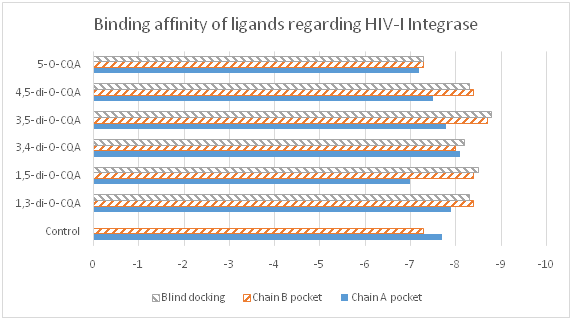
Fig. 4 Binding affinity of ligands towards HIV-I integrase (kcal/mol) – more negative values are better.
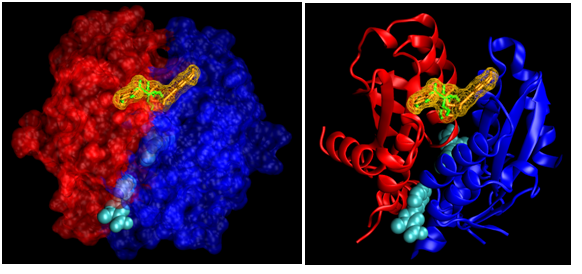
Fig. 5 a and b– New binding site for 3,4-diCQA (orange) and 5-CQA (green) on HIV-I integrase, Chain A (blue), Chain B (red), inhibitor from the crystal structure (cyan).
With this series of dockings it was possible to confirm several aspects of the works from Zhu et al.(16) and from Hu et al.(17). Firstly, the main finding of Zhu et al., was corroborated, that the inhibition of HIV-I integrase by diCQAs is performed in an allosteric site. The degree of inhibition is comparable to that of the control. This is corroborated even further by the fact that when the CQAs were docked to the active site region of HIV-I integrase described by Sotriffer & McCammon (19) the affinity of all CQAs is quite low (from -5.7 to -5.2 kcal/mol – data not shown).
It is clear that naturally occurring CQAs are poor inhibitors of HIV-I protease and this is likely due the fact that they do not contain any macro cycles or resemble the conformation of a β-strand protein. The results from the blind docking also support this as all molecules were docked with a very similar conformation at the active site (data not shown).
Naturally occurring caffeoylquinic acids do not appear to be suitable for inhibiting HIV-I protease but an etherification reaction between phenols should create a macro-cycle similar to current protease inhibitors.
HIV-I integrase is inhibited by naturally occurring diCQAs especially 3,4 and 3,5-diCQA. A new possible binding site for the 3,4-diCQA and 5-CQA was found between chain A and B. It is possible that, by binding here, the CQAs disrupt the connection between the chains of the integrase leading to a loss of activity.
Considering the different results obtained in the several dockings that were performed in this work and the results from Hu et al.,(17) the best course of action to determine where diCQAs bind in HIV-I integrase would be to solve its crystal structure bound to several CQAs. This, of course, would be a challenge in itself as the crystal structure of the full-length HIV-I integrase and its complexes with DNA has not been completely solved yet.
The Authors would like to acknowledge the Portuguese Foundation for Science and Technology (FCT) for the strategic project PEst-OE/QUI-674/UI0674/2014.
Tunnicliffe JM, Shearer J. Coffee, glucose homeostasis, and insulin resistance: physiological mechanisms and mediators. Appl Physiol Nutr Metab. 2008 Dec;33(6):1290–300. PMid:19088791
View Article PubMed/NCBIHenry-Vitrac C, Ibarra A, Roller M, Mérillon J-M, Vitrac X. Contribution of chlorogenic acids to the inhibition of human hepatic glucose-6-phosphatase activity in vitro by Svetol, a standardized decaffeinated green coffee extract. J Agric Food Chem. 2010 Apr 14;58(7):4141–4. PMid:20302380
View Article PubMed/NCBILee WJ, Zhu BT. Inhibition of DNA methylation by caffeic acid and chlorogenic acid, two common catechol-containing coffee polyphenols. Carcinogenesis. 2006 Feb;27(2):269–77. PMid:16081510
View Article PubMed/NCBIBelkaid A, Currie J-C, Desgagnés J, Annabi B. The chemopreventive properties of chlorogenic acid reveal a potential new role for the microsomal glucose-6-phosphate translocase in brain tumor progression. Cancer Cell Int. 2006 Jan;6(7). PMid:16566826
PubMed/NCBISuzuki A, Kagawa D, Ochiai R, Tokimitsu I, Saito I. Green coffee bean extract and its metabolites have a hypotensive effect in spontaneously hypertensive rats. Hypertens Res. 2002 Jan;25(1):99–107. PMid:11924733
View Article PubMed/NCBIOchiai R, Jokura H, Suzuki A, Tokimitsu I, Ohishi M, Komai N, et al. Green coffee bean extract improves human vasoreactivity. Hypertens Res. 2004 Oct;27(10):731–7. PMid:15785008
View Article PubMed/NCBIKönig B, Dustmann JH. The caffeoylics as a new family of natural antiviral compounds. Naturwissenschaften. 1985 Dec;72(12):659–61. PMid:4088317
View Article PubMed/NCBINeamati N, Hong H, Sunder S, Milne GW, Pommier Y. Potent inhibitors of human immunodeficiency virus type 1 integrase: identification of a novel four-point pharmacophore and tetracyclines as novel inhibitors. Mol Pharmacol. 1997 Dec;52(6):1041–55. PMid:9415714
View Article PubMed/NCBIMcDougall B, King PJ, Wu BW, Hostomsky Z, Reinecke MG, Robinson WE. Dicaffeoylquinic and dicaffeoyltartaric acids are selective inhibitors of human immunodeficiency virus type 1 integrase. Antimicrob Agents Chemother. 1998 Jan;42(1):140–6. PMid:9449274
PubMed/NCBIKing PJ, Ma G, Miao W, Jia Q, McDougall BR, Reinecke MG, et al. Structure-activity relationships: analogues of the dicaffeoylquinic and dicaffeoyltartaric acids as potent inhibitors of human immunodeficiency virus type 1 integrase and replication. J Med Chem. 1999 Feb 11;42(3):497–509. PMid:9986720
View Article PubMed/NCBIMa C-M, Kawahata T, Hattori M, Otake T, Wang L, Daneshtalab M. Synthesis, anti-HIV and anti-oxidant activities of caffeoyl 5,6-anhydroquinic acid derivatives. Bioorg Med Chem. Elsevier Ltd; 2010 Jan 15;18(2):863–9. PMid:20004585
View Article PubMed/NCBIDe Clercq E. Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV. Int J Antimicrob Agents. 2009 Apr;33(4):307–20. PMid:19108994
View Article PubMed/NCBICalugi C, Guarna A, Trabocchi A. Identification of constrained peptidomimetic chemotypes as HIV protease inhibitors. Eur J Med Chem. Elsevier Masson SAS; 2014 Sep 12;84:444–53.
Chua KCH, Pietsch M, Zhang X, Hautmann S, Chan HY, Bruning JB, et al. Macrocyclic protease inhibitors with reduced Peptide character. Angew Chem Int Ed Engl. 2014 Jul 21;53(30):7828–31. PMid:24903745
View Article PubMed/NCBIWensing AM, Calvez V, Günthard HF, Johnson V a, Paredes R, Pillay D, et al. 2014 update of the drug resistance mutations in HIV-1. Top Antivir Med. 2014;22(3):642–50. PMid:25101529
PubMed/NCBIZhu K, Cordeiro M, Atienza J. Irreversible inhibition of human immunodeficiency virus type 1 integrase by dicaffeoylquinic acids. J Virol. 1999;73(4):3309–16. PMid:10074185
PubMed/NCBIHu Z, Chen D, Dong L, Southerland W. Prediction of the interaction of HIV-1 integrase and its dicaffeoylquinic acid inhibitor through molecular modeling approach. Ethn Dis. 2010;20:1–10.
Trott O, Olson AJ. AutoDock Vina. J Comput Chem. 2010;31(2):445–61.
Sotriffer CA, McCammon JA. Active Site Binding Modes of HIV-1 Integrase Inhibitors. J Med Chem. American Chemical Society; 2000 Nov;43(22):4109–17.