Prof Dr Puis T Mpiana
Email: ptmpiana@gmail.com
© 2019 Sift Desk Journals. All Rights Reserved
VOLUME: 5 ISSUE: 1
Prof Dr Puis T Mpiana
Email: ptmpiana@gmail.com
Aristote Matondo1, Jason T Kilembe1, Domaine T Mwanangombo1, Beaudrique M Nsimba1, Benjamin Z Gbolo2,3, Gedeon N Bongo2,3, Koto-te-Nyiwa Ngbolua2,3, Dorothée D Tshilanda1, Damien S T Tshibangu1, Virima Mudogo1, Pius T Mpiana1*
1Department of Chemistry, Faculty of Sciences, University of Kinshasa, P.O Box 190, Kinshasa XI, Democratic Republic of the Congo
2Department of Biology, Faculty of Sciences, University of Kinshasa, P.O Box 190, Kinshasa XI, Democratic Republic of the Congo
3Department of Basic Sciences, Faculty of Medicine, University of Gbado-Lite, P.O Box 111, Gbado-Lite, Democratic Republic of the Congo
Puis Tshimankinda Mpiana, Facing COVID-19 via anti-inflammatory mechanism of action: Molecular docking and Pharmacokinetic studies of six anti-inflammatory compounds derived from Passiflora edulis(2021)Journal of Computational Chemistry & Molecular Modeling 5(1)
SARS-CoV-2 is the causative agent of the COVID-19 disease. Pathophysiologically, high levels of proinflammatory cytokines in the serum of SARS-CoV-2 patients are reported, which is so-called the cytokine storm. In this study, molecular docking calculations of six bioactive compounds from Passiflora edulis with anti-inflammatory activity in interaction with the main protease of SARS-CoV-2 were performed, and their pharmacokinetic properties were predicted. The results of their molecular simulations and the ADME-T profiles of each ligand (Absorption, Distribution, Metabolism, Excretion and Toxicity) suggest their use as potential treatment for SARS-CoV-2. Among the six investigated compounds in which four flavonoids and two alkaloids, the best docked ligands are quercetin (-8.2 kcal/mol), chrysin (-8.0 kcal/mol), kaempferol (-7.9 kcal/mol) and luteolin (-7.7 kcal/mol), both flavonoids compounds. Their pharmacokinetic studies using SwissADME, preADMET and pkCSM Web servers establish the good ADMET profile for each ligand.
Keywords: COVID-19, SARS-CoV-2, Cytokine storm, Passiflora edulis, anti-inflammatory activity, ADMET properties
Other than vaccine development, people around the world are waiting for the famous news from researchers: a molecule against the Severe Acute Respiratory Syndrome 2 (SARS-CoV-2) has been found. The SARS-CoV-2 is the causative agent of the novel ß-Coronavirus (2019-nCoV) or the Corona Virus Disease (COVID-19) that is a pneumonia infection characterized by the hyperproduction of mainly proinflammatory cytokines (IL-1, IL-6, TNF-α, etc) (Costela-Ruiza et al, 2020). In the active research of finding molecule that can treat COVID-19, two approaches are currently being used. One is to find molecules that can be used as potential treatment against COVID-19 among several FDA-approved drugs (Chandel et al, 2020; Contini, 2020), while the other is to identify from plants biodiversity potential inhibitors (phytochemicals) of SARS-CoV-2’s main protease using molecular modeling approaches (Gyebi et al, 2020; Mpiana et al, 2020).
During molecular modeling approaches of finding potentials inhibitors, particular emphasis is placed on the significance of binding affinity of ligand-protein complexes and on their drug-likeness properties (Gyebi et al, 202; Kar et al, 2020). However, it should be mentioned that the biological activities of these molecules are as well very important (Kandeel and Al-Nazawi, 2020). Further, the most common trend is that an anti-COVID-19 molecule might be derived from a plant endowed with antiviral properties (Mbadiko et al, 2020; Boopathi et al, 2019; Ngbolua et al, 2020; Aouidate et al, 2020). Nevertheless, what is abundantly clear in this moment is that the most important cause of COVID-19 related deaths is respiratory failure which is due to pneumonia (an acute inflammatory lung injury), which itself varies depending on the disease severity level, but also alveolar damage that can precipitate acute respiratory distress syndrome (ARDS) (Zhang et al, 2020). The innate immune response is then to produce pro-inflammatory cytokines and chemokines to contain and stop the infection. Pathophysiologically, previous studies have reported high levels of various cytokines (the so-called cytokine storm) and chemokines in the serum of SARS-CoV-2 patients (Cron and Chatham, 2020; Diao et al, 2020). In addition, Fidan and Aydoğdu recently reported that various pro-inflammatory cytokines such as IL-6, IL-1, the tumor necrosis factor (TNF-α) induce a migration of leukocytes into lungs, that then secrete the reactive oxygen species and proteases that damage capillary endothelium and alveolar epithelium (Fidan and Aydoğdu, 2020).
Based on the relevant clinical characteristics, phytocompounds derived from Passiflora edulis whose isolated molecules have several therapeutic properties such as anti-inflammatory, antioxidant, anti-microbial, anti-cancer… can be used for the treatment of COVID-19 as supported in the following lines:

Fig.1. Leaves and Flowers (left) and yellow passion fruits (right) of P. edulis
2.1. Literature review
Based on information reported above, six phytochemicals derived from P. edulis with anti-inflammatory activity are chosen for this study. Chrysin or 5,7-dihydroxyflavone (5,7-dihydroxy-2-phenyl-4H-chromen-4-one), kaemferol or 3,5,7-trihydroxy-2-(4-hydroxyphenyl)-4H-1-benzopyran-4-one), luteolin or 2-(3,4-dihydroxyphenyl)-5,7-dihydroxychromen-4-one, quercetin or 2-(3,4-dihydroxyphenyl)-3,5,7-trihydroxychromen-4-one, harmol or 1-Methyl-2,9-dihydropyrido[3,4-b]indol-7-one and harmine or 7-Methoxy-1-methyl-9H-pyrido[3,4-b] indole. The four first compounds are flavonoids while the two latter compounds are alkaloids. Chemical structures of compounds were retrieved from literature sources. Their 2D structures have been sketched using Marvin JS and their 3D structures retrieved from PubChem/NLM. Bibliographical references were made using a bibliographical software "Mendeley".
2.2. Molecular Docking
The structure of the 3-Chymotrypsin-Like protease (3CLpro) or the COVID-19 virus main protease (Mpro) which is among the most studied SARS-CoV-2 proteases was obtained from PDB (Protein Data Bank) database (PDB ID: 2GTB) and imported into chimera for visualizing the binding domain of the complex and identifying the amino acids in the binding pocket as well. The hydrogen atoms were added to the protein in order to correct the ionization and tautomeric states of the amino acid residues. Furthermore, the water molecules and complexes bound to receptor molecule were removed before the docking. Incomplete side chains were replaced using Drunbrack rotamer library. In addition, the protein was subjected to energy minimization by applying the AMBER 14SB force field, and AM1-BCC was used for other residues with a maximum number of 200 steps at RMS gradient of 0.02. The optimized protein was saved in pdbqt format and imported to PyRx for molecular docking which was carried out by means of Autodock Vina virtual screening tool (Trott and Olson, 2010). The validation of the docking study was performed by re-docking the reference ligand into an appropriate protein cavity. Re-docking is accepted if the root mean square value (RMSD) < 2.0 Å. Figure 2 displays schematic structure of the SARS-CoV-2 Mpro/3CLpro (a) and the complex formed between the SARS-CoV-2 Mpro and 2GTB as a potential drug target for the new coronavirus-2 (b). According to Xu and co-workers, 2GTB is the main protease found in the coronavirus associated with the severe acute respiratory syndrome (SARS), and that the main protease in 2019-nCoV shares 96% similarity with that in SARS (Xu et al, 2020).
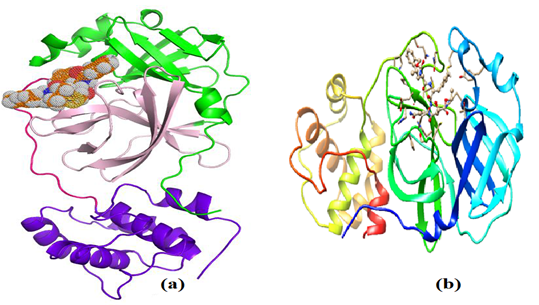
Figure 2: Schematic structure of the SARS-CoV-2 Mpro (a) and the complex formed between the SARS-CoV-2 Mpro and co-crystallized inhibitor 2GTB (b)
2.2.1. Generation of ligand dataset and pharmacokinetic profiles
The selected compounds derivatives from various literature resources (Zucolotto et al, 2012; He et al, 2020; ) were drawn using Marvin JS. Figure 3 shows the 3D structures of the sketched compounds retrieved from PubChem/NLM. The 3D ligands were then saved in .sdf format. Ligands optimization was performed by using universal force field (UFF) with conjugate gradients algorithm of 200 Steps, and then analyzed for pharmacokinetic properties. Bioinformatics resources have been employed in the prediction of ADME properties (Absorption, Distribution, Metabolism and Excretion) using the SwissADME database (Daina et al, 2017). During the early stages of drug discovery, the ligand to be selected as a hit must be non-carcinogenic and non-hepatotoxic. The toxicity assessment (ADMET, T for Toxicity) that allows to predict the mutagenicity (Ames test) and carcinogenicity of the potential ligands was made using the preADMET server, Korea (Lee et al, 2004), while the hepatotoxicity and the oral rat acute toxicity were assessed using the pkCSM server (Pires et al, 2015).
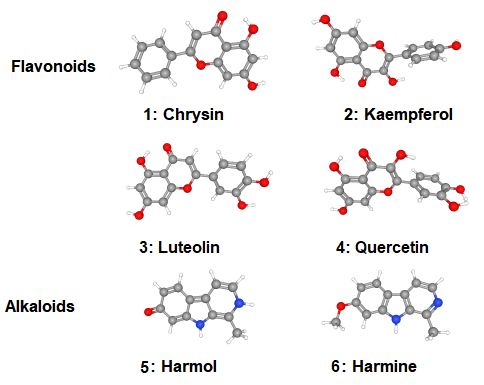
Figure 3: 3D Structures of selected flavonoids and alkaloids compounds 1–6 derived from P. edulis.
3.1. Energetics and geometries
Noncovalent interactions, mainly H-bonds (Matondo et al, 2019), van der Waals and π-π interactions (stacked/parallel and T-shaped/perpendicular conformations) (Kasende et al, 2017) are forces that drive and determine the binding of ligand-protein interactions. The most common tool to evaluate the strength of binding between ligand-protein interactions is molecular docking. The docking results obtained using AutoDock Vina virtual screening tool between ligands 1-6, the native or reference ligand with the SARS-CoV-2’s main protease (Mpro or 3CLpro) are gathered in Table 1.
Table 1: Binding affinity (kcal/mol) of 2GTB and Ligands 1-6 with SARS-CoV-2 Mpro.
|
Receptor PDB ID |
Ligands |
Binding Affinity (ΔG in Kcal/mol) |
|
2GTB |
Lopinavir |
-8.4 |
|
Nelfinavir |
-8.1 |
|
|
1 |
-8.0 |
|
|
2 |
-7.9 |
|
|
3 |
-7.7 |
|
|
4 |
-8.2 |
|
|
Ref. Ligand |
-7.4 |
|
|
5 |
-6.7 |
|
|
6 |
-6.4 |
Since Lopinavir and Nelfinavir, two FDA approved drugs for the treatment of human immunodeficiency virus (HIV)/acquired immunodeficiency syndrome patients can represent potential treatment options (Khaerunnisa et al, 2020), they were used as drug standards for comparison.
The binding affinity values of 6 ligands ranging from -6.40 to -8.22 kcal/mol place the four flavonoids compounds as the best docked ones to the SARS-CoV-2 main protease. The most strongly bound to the protease cavity is ligand 4 or quercetin (-8.22 kca/mol), followed by ligand 1 or chrysin (-8.04 kcal/mol). The overall trend of complexes stability follows the pattern: ligand 4 > ligand 1> ligand 2 > ligand 3> ligand 5> ligand 6>. Based on data presented in this table, the binding energies of the four flavonoids compounds are somewhat close to those of Lopinavir/Nelfinavir and higher than that of the reference ligand. However, as part of principles that drive drug discovery, this does not mean that ligands 1-6 can automatically inhibit the virus action, or ligands 5 and 6 cannot be considered as hits, without establishing the pharmacokinetic properties of each ligand.
Turning next to the types of noncovalent interactions established between ligands and the SARS-CoV-2 Mpro, one can see in figure 4 below that the complexes are mainly stabilized by hydrogen bonding interactions, but also supported by van der walls and π/π interactions. At this stage, the stability of ligands 1-4 which are flavonoids compounds over ligands 5 and 6 can be explained by the presence of multiple OH groups that can be act simultaneously as hydrogen bonds acceptors (HBA) and donors (HBD) (Kasende et al, 2016).
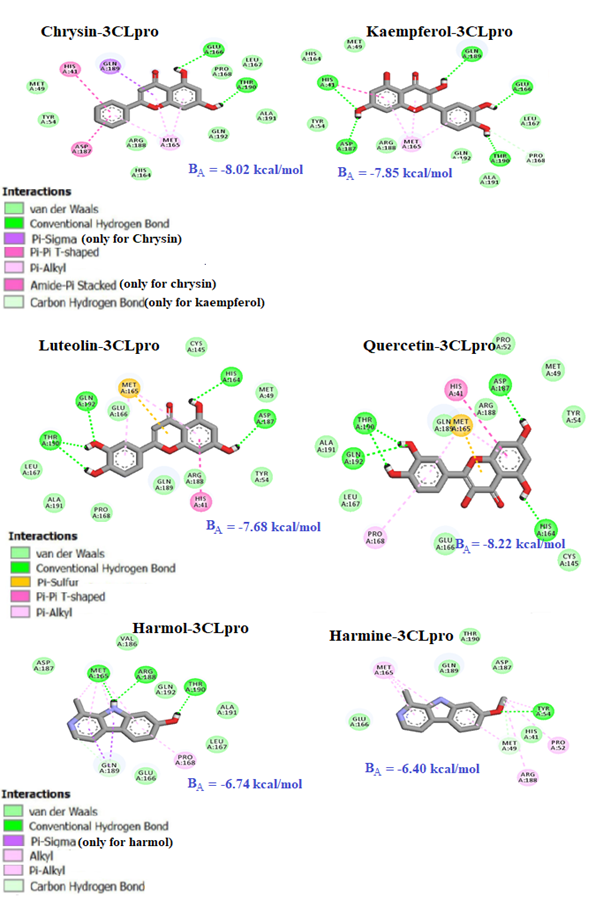
Fig. 4. Interaction map of ligands 1-6 with the main protease of coronavirus 3CLpro
In addition, the presence of three aromatic rings in flavonoids compounds offer much possibilities to π-π interactions to take place. Such interactions are mainly stabilized by dispersion or van der Waals forces, important in the ligand-protein interactions (Kasende et al, 2017).
H-bonds parameters (distances and angles) between the protein target and ligands 1-6 along with the involved groups (ligands) and the amino acids residues of the Mpro engage in H-bonding interaction are summarized in Table 2.
Table 2. Hydrogen-bonds parameters derived from docking of ligands 1-6 with SARS-CoV-2 Mpro
|
Ligand |
AA residues |
Ligand group |
δ (Å) |
θ(˚) |
|
1 |
GLU166 |
O-H |
1.99 |
155 |
|
HIS189 |
O-H |
2.17 |
163 |
|
|
THR180 |
O-H |
2.01 |
142 |
|
|
ASP187 |
O-H |
2.18 |
140 |
|
|
HIS41 |
O=C2 |
2.17 |
153 |
|
|
2 |
HIS41 |
O-H |
2.30 |
163 |
|
ASP187 |
O-H |
2.29 |
135 |
|
|
GLU166 |
O-H |
1.86 |
153 |
|
|
THR190 |
O=C2 |
2.24 |
158 |
|
|
GLN189 |
O-H |
1.97 |
160 |
|
|
3 |
THR190 |
O-H |
2.15 |
145 |
|
THR190 |
O-H |
2.15 |
150 |
|
|
GLN192 |
O-H |
2.36 |
144 |
|
|
HIS164 |
O-H |
2.32 |
130 |
|
|
ASP187 |
O-H |
2.00 |
170 |
|
|
4 |
GLN192 |
O-H |
2.10 |
144 |
|
THR190 |
O-H |
2.20 |
155 |
|
|
THR190 |
O-H |
2.22 |
150 |
|
|
HISP164 |
O-H |
1.99 |
140 |
|
|
ASP187 |
O-H |
1.78 |
165 |
|
|
5 |
ARG188 |
H-N |
2.06 |
145 |
|
THR190 |
H-O |
1.83 |
165 |
|
|
MET165 |
H-N |
2.04 |
151 |
|
|
6 |
TYR54 |
H-N |
2.18 |
160 |
It can be seen that ligands 1, 2, 3 and 4 form five conventional hydrogen bonds with the active site of the SARS-CoV-2 main protease, whereas the two weakest complexes are three and one hydrogen bonds for ligand 5 and ligand 6, respectively. The strongest (shortest) hydrogen bonding interaction is established between the residue of the amino acid ASP187 of the Mpro with the O-H group of the quercetin ligand (1.78 Å). The two adducts form the strongest complex.
3.2 Physicochemical properties and ADME-T profiles
Physicochemical property is an important parameter of a molecule that influences efficacy, safety or metabolism which could be predicted by using Lipinski’s rule of five (RO5) that is: molecular mass < 500; Hydrogen-bond donors (HBD) ≤ 5; Hydrogen-bond acceptors (HBA) < 10; and Log P < 5 (Lagorce et al, 2017). Prediction of in silico physicochemical parameters of the 6 ligands are grouped in Table 3.
Inspection of Table 3 shows that all ligands meet every single criterion of Lipinski’s rule of five and thus fully obey the rule. Consequently, all the investigated ligands are predicted to be easily absorbed and have good permeability and bioavailability. According to Ghose and co-workers, the molecular refractivity is a ubiquitous parameter for a drug molecule that cannot exceed 130 m3.mol-1 and not to be under 40 m3.mol-1 (Ghose et al, 1999).
Table 3. Predicted in silico physicochemical parameters using SwissADME online tool
|
Ligand |
Formula |
MW (Da) |
Log P |
HBD |
HBA |
PSA (Å) |
Refractivity (m3.mol-1) |
Violations |
Log S |
|
1 |
C15H10O4 |
254.24
|
2.27
|
2
|
4
|
70.67
|
71.97
|
0
|
-4.19 |
|
2 |
C15H10O6 |
286.24 |
1.70
|
4
|
6
|
111.13
|
76.01
|
0
|
-3.31
|
|
3 |
C15H10O6 |
286.24 |
1.86 |
4 |
6 |
117.31 |
76.01 |
0 |
-3.71 |
|
4 |
C15H10O7 |
302.24 |
1.63 |
5 |
7 |
131.36 |
78.03 |
0 |
-3.16 |
|
5 |
C12H10N2O |
198.22 |
1.68 |
2 |
1 |
48.65 |
61.39 |
0 |
-2.18 |
|
6 |
C13H12N2O |
212.25 |
2.07 |
1 |
2 |
37.91 |
65.06 |
0 |
-4.05 |
With MW= Molecular weight, Log P = Lipophilicity, PSA = Polar Surface Area, Log S = water solubility
None violation is observed here for all the investigated ligands as can be seen in Table 3. Finally, as pointed out by Cerqueira and co-authors, for optimal drug absorption and distribution, the polar surface area (PSA) values cannot be higher than 140 Å (Cerqueira et al, 2015). Once again, none violation is observed here. Three potential candidates for the inhibition of the SARS-CoV-2 3CLpro have PSA values almost two to three times less than the recommended value (ligands 1, 5 and 6), while ligands 2, 3 and 4 have PSA value higher than 100 Å.
The next step to deal with is to establish the ADME/T profiles of each ligand. In fact, a major issue after identifying stables complexes, that is, lead or hit compounds, is to evaluate their ADME parameters and cardiotoxicity.
These pharmacokinetic properties are very important parameters in the computer-aided drug discovery since they allow one to retract some hit from early-stage trials. The ADME properties are evaluated by using SwissADME and pkCSM servers, but other parameters such as the Blood-Brain Barrier (BBB), the Human Intestine Absorption (HIA) and the skin permeability come from the preADMET server. The selected endpoints for toxicity are Ames test and Rodent Carcinogenicity (rat) in preADMET server, hepatotoxicity and oral rat acute toxicity (LD50) in pkCSM server. These parameters are gathered in Table 4.
Table 4. ADME-T profile of ligands 1-6.
|
Parameter |
Ligand 1 |
Ligand 2 |
Ligand 3 |
Ligand 4 |
Ligand 5 |
Ligand 6 |
|
Absorption & Distribution |
|
|
|
|
|
|
|
BBB |
0.933 |
0.286 |
0.368 |
0.173 |
0.320 |
3.798 |
|
HIA (%) |
92.644 |
79.439 |
81.132 |
77.207 |
94.263 |
92.827 |
|
Skin permeability (log Kp) |
-3.346 |
-4.323 |
-4.280 |
-4.433 |
-4.662 |
-4.386 |
|
Bioavailability score |
0.55 |
0.55 |
0.55 |
0.55 |
0.55 |
0.55 |
|
Metabolism |
|
|
|
|
|
|
|
CYP2D6 |
No |
No |
No |
No |
No |
Yes |
|
CYP3A4 |
Yes |
No |
No |
No |
No |
Yes |
|
Excretion |
|
|
|
|
|
|
|
Total clearance |
0.48 |
0.50 |
0.50 |
0.41 |
0.59 |
0.62 |
|
Renal OCT2 substrate |
No |
No |
No |
No |
No |
No |
|
Toxicity |
|
|
|
|
|
|
|
Ames test |
Yes |
No |
No |
No |
Yes |
Yes |
|
Hepatotoxicity |
No |
No |
No |
No |
No |
No |
|
Carcinogenicity (rat) |
Negative |
Positive |
Negative |
Negative |
Negative |
Negative |
|
oral rat acute toxicity (LD50, in mol/kg) |
2.486 1243 |
2.197 1099 |
2.455 1228 |
2.471 1236 |
2.781 1391 |
2.999 1450 |
Values in bold are expressed in mg/kg
According to the binding affinity values, it was derived the following decreasing order in the complexes formed between ligands and the SARS-CoV-2 3CLpro or Mpro: Ligand 4 > Ligand 1 > Ligand 2 > Ligand 3> Ligand 5 > Ligand 6. This order only reflects the thermodynamic stability of complexes. However, the stability over time of the ligand in a protein interaction site depends on other factors. For a ligand to be used for therapeutic purposes, its absorption, distribution, metabolism, excretion and toxicity are aspects to take into account. To pursue further, it is worthy to point out that first of all, such a ligand must be non-hepatotoxic and non-carcinogenic (Alamri, 2020).
Scrutiny of toxicity outcomes in Table 4 reveals that all potential ligands are non-hepatotoxic. With regards to the carcinogenicity, the results predict the carcinogenic activity only for the ligand 2. This encouraging result of toxicity assessment allows us to go back to ADME properties. The ability of a drug molecule to cross into the brain is an important propriety to improve the efficacy of drugs (reduce side effects and toxicities). The BBB values for the potential candidates are all positive and the lowest value is found in ligand 4 (0.173) which forms the strongest complex with the SARS-CoV-2 main protease. The probability of intestinal absorption by human is very high, and in the other hand almost the same for ligands 1, 5 and 6; and on the other hand almost the same for ligands 2, 3 and 4. Ligand 4 has the smallest probability (79.44 %) of being absorbed by human intestine, in contrast with its binding affinity with the COVID-19 protease. The recommended value of the skin permeability or log Kp for a drug molecule is set at more than -2.5 cm/h. Interestingly, the computed log Kp values range from -3.3 to -4.7 cm/h. Finally, the bioavailability score which is evaluated to 0.55 confirms that ligands 1-6 have good absorption and distribution since all potential candidates may have more than 10% of bioavailability in rat (Martin, 2015).
The Cytochrome P450 inhibition as metabolic indicators including CYPs: 1A2, 2C19, 2C9, 2D6 and 3A4 are predicted. Nevertheless, only CYP2D6 and CYP3A4 are responsible for drug metabolism (Barbosa et al, 2008; Dofferhoff et al, 2020). Interestingly, the three best candidates according to their binding affinity are found to be non-inhibitors of CYP2D6 and CYP3A4 except hits 1 and 6 that affect the CYP3A4, and hit 6 which in addition affects the CYP2D6. This result rules the ligand 6 out from the list of potential candidates for the inhibition of the SARS-CoV-2 main protease, a result which is moreover in good agreement with its lower free enthalpy (-6.40 kcal/mol).
Turning next to excretion also called elimination, the total clearance is directly linked to the renal OCT2 (organic cation transporter 2) substrate that offers helpful information on potential contraindications. The selected 4 flavonoids and 2 alkaloids compounds are predicted to be not renal OCT2 substrates. This means that all the 6 phytocompounds can be eliminated through the OCT2 substrate. Surprisingly, the total clearance values of the investigated compounds vary almost inversely with their binding affinities values. In addition, ligands 2 and 3 which have the same molecular weight of both chemical formula C15H10O6 have exactly the same total clearance.
Closing finally the ADME-T profiles of the six compounds, their oral acute toxicity (LD50) are classified in category or class 4, meaning that they are slightly toxic (Globally Harmonized System: 300 < Category 4 ≤ 2000) and can thus be considered as safe.
Bacteria, fungi, viruses can cause serious infections and diseases in the world. Some of these infections and diseases are curable while other are not. Even for those that are curable, there is sometimes multiple drug resistance. Since humanity exists, plants have been one of man’s best friends. While it is obvious that plants are a source of vitamins, proteins, minerals, they are as well source of phytocompounds containing excellent therapeutic properties: antioxidant, anti-inflammatory, antifungal, antimicrobial, antitumor activities. Passiflora edulis, also known as passion fruit, passion flower, purple granadilla, or “maracuja” (DR Congo or Brazil), is widely cultivated for its edible fruit. With numerous biological activities, the passion fruit also contains vitamins A, C, E, K, minerals such as Zn (0.10 g), Mg (29 mg), K (348 mg), Ca (12 mg), etc (Barbosa et al, 2008). On one hand, vitamins A and K could help to fight the COVID-19 (Dofferhoff et al, 2020). On the other hand, these chemical elements mainly Zn, although indispensable as enzymatic co-factors, a slight increase in their intracellular concentration inhibits the replication of retroviruses including SARS-CoV-1 (te Velthuis et al, 2010) important in the management of COVID-19. Owing to its numerous therapeutic activities, P. edulis has traditional or ethno medicinal uses in many countries. Of complex phytochemistry, its secondary metabolites have numerous health benefits and very recently, as stated above, Jabareen and co-workers reported in an experimental study the antiviral activity of P. edulis leaves on some viruses’ species including Herpes Simplex Virus Type 1 and 2, Varicella-Zoster Virus, etc (Jabareen et al, 2013). Since P. edulis exhibits anti-inflammatory capabilities essential to stem the cytokine storm, this study is conducted in order to identify potential inhibitors from a set of 6 phytochemicals endowed with anti-inflammatory activity. The 6 selected compounds (four flavonoids and two alkaloids) reacted with the SARS-CoV-2 3CLpro or Mpro, and an order of thermodynamic stability was obtained.
Compared with Lopinavir and Nelfinavir that are protease inhibitors recommended for the treatment of SARS and MERS, the binding affinities of the four top compounds, both flavonoids, are very close to those of two anti-HIV drugs, and even a bit higher to that of the ligand reference. Indeed, the ligand 1 or chrysin has anti-inflammatory, antibacterial and antioxidant activities (Modi et al, 2018; Abotaleb et al, 2019), while ligand 2 or kaempferol exhibits antitumor, antioxidant and anti-inflammatory capabilities (Wang et al, 2018). In fact, our previous studies showed that aloe vera represents potential treatment for COVID-19 (Mpiana et al, 2020), and three of its phytochemicals were identified as potential inhibitors of SARS-CoV-2 main protease, in which two compounds exhibit anti-inflammatory effect (Mpiana PT et al, 2020). The anti-inflammatory activity of luteolin in experimental animal models was reported by Ziyan and co-workers (Ziyan et al, 2007), and recently, a study by Lesjak and co-authors showed antioxidant and anti-inflammatory activities of quercetin and its derivatives (Lesjak et al, 2018).
One can remember that the hyperproduction of proinflammatory cytokines is the main reason that causes morbidity and mortality in SARS-CoV-2 patients. Thus, the application of anti-inflammatory molecules is a mechanistically-sound strategy for treatment development (Brennecke et al, 2020). The established ADME-T profile of each ligand suggests that the four flavonoids (chrysin, kaempferol, luteolin and quercetin) might be used as potential treatment of SARS-CoV-2.
In conclusion, the strategy adopted in this work consisted in exploring the inhibitory power of six phytochemicals derived from P. edulis. Given the cytokine storm, six compounds exhibiting anti-inflammatory activity, among which four flavonoids and two alkaloids, were each paired with the SARS-CoV-2 main protease in order to evaluate first their thermodynamic stability. The docking affinity scores showed that ligands 1-4 (flavonoids) are more stables than ligands 5 and 6 (alkaloids). Then, the Lipinski’s rule of five and the pharmacokinetic studies using SwissADME, preADMET and pkCSM showed that these phytochemicals have good ADME-T profiles, mainly the flavonoids compounds. Consequently, chrysin, kaempferol, luteolin and quercetin as the four top compounds can be used via anti-inflammatory mechanism of action to fight the overproduction of proinflammatory cytokines in SARS-CoV-2 patients.
Costela-Ruiza VJ, Illescas-Montesa R, Puerta-Puerta JM et al (2020) SARS-CoV-2 infection: The role of cytokines in COVID-19 disease. Cytokine and Growth Factor Reviews.
View ArticleChandel V, Raj S, Rathi B, Kumar D (2020) In silico identification of potent COVID-19 main protease inhibitors from FDA approved antiviral compounds and active phytochemicals through molecular docking: A drug repurposing approach. Preprints.
Contini A (2020) Virtual screening of an FDA approved drugs database on two COVID-19 coronavirus proteins. ChemRxiv Preprint.
View ArticleGyebi GA, Ogunro OB, Adegunloye AP et al (2020) Potential Inhibitors of Coronavirus 3-Chymotrypsin Like Protease (3CLpro): An in silico screening of Alkaloids and Terpenoids from African medicinal plants. J Biomol Struct Dyn 1-13.
Mpiana PT, Ngbolua KN, Tshibangu DST et al (2020) Identification of potential inhibitors of SARS-CoV-2 main protease from Aloe vera compounds: A molecular docking study. Chem Phys Lett 754:137751.
View ArticleKar P, Sharma NR, Singh B et (2020) Natural compounds from Clerodendrum spp. as possible therapeutic candidates against SARS-CoV-2: An in silico investigation. J Biomol Struct Dyn.
View ArticleKandeel M, Al-Nazawi M (2020) Virtual screening and repurposing of FDA approved drugs against COVID-19 main protease. Life Sciences 257:117627.
Mbadiko CM, Inkoto CL, Gbolo BZ et al (2020) A Mini Review on the Phytochemistry, Toxicology and Antiviral Activity of Some Medically Interesting Zingiberaceae Species. J Complem Altern Medic Res 9 (2020) 44-56. .
View ArticleBoopathi S, Poma AB, Kolandaivel P (2020) Novel 2019 coronavirus structure, mechanism of action, antiviral drug promises and rule out against its treatment. J Biomol Struct Dyn.
View ArticleNgbolua KN, Mbadiko CM, Matondo A et al (2020) Review on Ethno-botany, Virucidal Activity, Phytochemistry and Toxicology of Solanum genus: Potential Bio-resources for the Therapeutic Management of Covid-19. Eur J Nutrition Food Safety 12:35-48. .
View ArticleAouidate A, Ghaleb A, Chtita S et al (2020) Identification of a novel dual-target scaffold for 3CLpro and RdRp proteins of SARS-CoV-2 using 3D-similarity search, molecular docking, molecular dynamics and ADMET evaluation. J Biomol Struct Dyn (2020).
View ArticleZhang JJ, Dong X, Cao YY et al (2020) Clinical characteristics of 140 patients infected with SARS-CoV-2 in Wuhan, China. Allergy.
View ArticleCron RQ, Chatham WW. The Rheumatologist’s Role in Covid-19. The Journal of Rheumatology.
View ArticleDiao B, Wang C, Wang R et al (2020) Human Kidney is a Target for Novel Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Infection. MedRxiv. .
View ArticleFidan C, Aydoğdu A (2020) As a potential treatment of COVID-19: Montelukast, Medical Hypothesis (2020) doi: .
View ArticleMontanher AB, Zucolotto SM, Schenkel EP, Fröde TS (2007) Evidence of anti-inflammatory effects of Passiflora edulis in an inflammation model. J Ethnopharmacology 109 (2007) 281–288.
Ledford H (2020) Coronavirus breakthrough: dexamethasone is first drug shown to save lives. Nature 582:469.
Cazarin CBB, da Silva JK, Colomeu TC et al (2015) Intake of Passiflora edulis leaf extract improves antioxidant and anti-inflammatory status in rats with 2,4,6-trinitrobenzenesulphonic acid induced colitis. J Funct Foods 17:575–586.
Cazarin CBB, Rodriguez-Nogales A, Algieri et al (2016) Intestinal anti-inflammatory effects of Passiflora edulis peel in the dextran sodium sulphate model of mouse colitis. J Funct Foods 26:565–576.
He X, Luan F, Yang Y et al (2020) Passiflora edulis: An Insight into Current Research on Phytochemistry and Pharmacology. Front. Pharmacol. 20:617-633.
Li SP, Wang YW, Qi SL et al (2018) Analogous b-carboline alkaloids harmaline and harmine ameliorate scopolamine induced cognition dysfunction by attenuating acetylcholinesterase activity, oxidative stress, and inflammation in mice. Front. Pharmacol. 9:346.
Liu X, Li M, Tan S et al (2017) Harmine is an inflammatory inhibitor through the suppression of NF-kB signaling. Biochem. Biophys. Res. Commun. 489:332–338.
Ingale AG, Hivrale AU (2010) Pharmacological studies of Passiflora sp. and their bioactive compounds, Afr. J. Plant Sci. 4:417-426.
Zas P, John S (2017) Phytochemical investigation and antioxidant activities of Passiflora edulis (Passion fruit) leaves from Ukhrul district, Manipur, India. World J. Pharm. Res. 6:793–801.
Jabareen A, Huleihil M, Huleihel M (2013) Effect of Extracts of Passiflora edulis Leaves on Herpes Viruses Infection. J. Virology Antiviral Res. 2:1-6.
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31:455–461.
Xu Z, Peng C, Shi Y et al (2020) Nelfinavir was predicted to be a potential inhibitor of 2019-nCoV main protease by an integrative approach combining homology modelling, molecular docking and binding free energy calculation. bioRxiv.
View ArticleZucolotto SM, Fagundes C, Reginatto FH et al (2012) Analysis of C-glycosyl Flavonoids from South American Passiflora Species by HPLC-DAD and HPLC-MS. Phytochem. Anal. 23:232–239.
Daina A, Olivier M, Zoete V (2017) SwissADME: a free web tool to evaluate pharmacokinetics, druglikeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7:42717.
Lee SK, Chang GS, Lee IH et al (2004) The preADME: PC-based program for batch prediction of ADME properties. EuroQSAR 9:5-10.
Pires DE, Blundell TL, Ascher DB (2015) pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using Graph- Based Signatures. J. Med. Chem. 58:4066–4072.
Matondo A, Thomas R, Tsalu PV et al (2019) α-methylation and α-fluorination electronic effects on the regioselectivity of carbonyl groups of uracil by H and triel bonds in the interaction of U, T and 5FU with HCl and TrH3 (Tr = B, Al). J. Mol. Graph. Model. 88:237–246.
View ArticleKasende OE, Matondo A, Muya JT, Scheiner S (2017) Interactions between temozolomide and guanine and its S and Se-substituted analogues, Int. J. Quantum Chem. 117:157−169.
View ArticleKhaerunnisa S, Hendra K, Rizki A et al (2020) Potential inhibitor of COVID-19 Main Protease (Mpro) from several medicinal plant compounds by molecular docking study.
View ArticleKasende OE, Matondo A, Muya JT, Scheiner S (2016) Interaction between temozolomide and HCl: preferred binding sites. Comput. Theor. Chem. 1075:82–86.
View ArticleLagorce D, Douguet D, Miteva MA, B.O.Villoutreix (2017) Computational analysis of calculated physicochemical and ADMET properties of protein-protein interaction inhibitors, Sci. Rep. 7:46277.
Ghose AK, Viswanadhan VN, Wendoloski JJ (1999) A knowledge- based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comp Chem. 1:55–68.
Cerqueira NM, Gesto D, Oliveira EF et al (2015). Receptor-based virtual screening protocol for drug discovery. Arch. Biochem. Biophys 582 (2015) 56–67.
Alamri MA (2020) Pharmacoinformatics and molecular dynamic simulation studies to identify potential small-molecule inhibitors of WNK-SPAK/OSR1 signaling that mimic the RFQV motifs of WNK kinases. Arab. J. Chem. 13:5107-5117.
View ArticleMartin YC (2015). A bioavailability score. J. Med. Chem. 48:3164–3170.
Barbosa PR, Valvassori SS, Bordignon Jr (2008) The aqueous extracts of Passiflora alata and Passiflora edulis reduce anxiety-related behaviors without affecting memory process in rats. J. Med. Food 11:282–288.
Dofferhoff ASM, Piscaer I, Schurgers LJ et al (2020) Reduced vitamin K status as a potentially modifiable prognostic risk factor in COVID-19. (2020). Preprints.
View Articlete Velthuis AJW, van den Worm SHE, Sims AC et al (2010) Zn2+ inhibits Coronavirus and arterivirus RNA polymerase activity in vitro and zinc ionophores block the replication of these viruses in cell culture, Plos Pathogens 6:1-10.
Modi F, Shailesh B, Patel J et al (2018) Evaluation of Pharmacokinetics, Antibacterial and Anti-Inflammatory Activities of Chrysin in Rat. Int. J. Current Microbiol. Appl. Sc. 7:1494-1503.
Abotaleb M, Samuel SM, Varghese E et al (2019) Flavonoids in Cancer and Apoptosis. Cancers (Basel) 11:28.
Wang J, Fang X, Ge L et al (2018) Antitumor, antioxidant and anti-inflammatory activities of kaempferol and its corresponding glycosides and the enzymatic preparation of kaempferol. PLoS One 13 (2018) e0197563.
Mpiana PT, Ngbolua KN, Tshibangu DST
Ziyan L, Yongmei Z, Nan Z et al (2007) Evaluation of the anti-inflammatory activity of luteolin in experimental animal models. Planta Med 73:221-226.
View ArticleLesjak M, Beara I, Simin N (2018) Antioxidant and anti-inflammatory activities of quercetin and its derivatives. J Funct Foods 40:68–75.
View ArticleBrennecke A, Villar L, Wang Z et al (2020) Is Inhaled Furosemide a Potential Therapeutic for COVID-19? Am. J. Med. Sc. (2020)
View Article