Sopi Thomas AFFI
Email: affithomas.sfa@univ-na.ci
© 2019 Sift Desk Journals. All Rights Reserved
VOLUME: 5 ISSUE: 3
Page No: 657-663
Sopi Thomas AFFI
Email: affithomas.sfa@univ-na.ci
Sopi Thomas AFFI*1, Doh SORO1, Souleymane COULIBALY2, Bibata KONATE1, Nahossé ZIAO1
1 Laboratoire de Thermodynamique et de Physico-Chimie du Milieu, Unité de Formation et de Recherche Sciences Fondamentales et Appliquées, Université Nangui Abrogoua, Abidjan, Côte d’Ivoire
2 Laboratoire de Constitution et Réaction de la Matière, Unité de Formation et de Recherche Sciences des Structures de la Matière et Technologie, Université Félix Houphouët-Boigny, Abidjan, Côte d’Ivoire
Sopi Thomas AFFI, Doh SORO, Souleymane COULIBALY, Bibata KONATE, Nahossé ZIAO, Modeling anticancer pharmacophore based on inhibition of HADAC7(2021)Journal of Computational Chemistry & Molecular Modeling 5(3) :657-663
Histone deacetylases (HDACs) are the target inhibition enzymes in cancer treatment via chemotherapy. Application of this therapeutic technique requires the use of drugs whose side effects are reduced and tiny with necessary safety. In this study, the methods and tools of pharmacophore modeling were used to investigate ten molecules known for their anticancer properties. Particular attention has been given to pinpoint a promising anti-cancer pharmacophore in order to lead new effective inhibitors. Using Discovery Studio 2.5 software, the ten compounds were docked within the active site of the HDAC7 enzyme. Analysis of the binding characteristics of all the compounds collected and tested in the model resulted in the characteristics produced by the 3D pharmacophore of the selected hypothesis. This led to note that the efficiency of any HDAC enzyme inhibitor was related to the characteristics of the designed pharmacophore. At the end, the pharmacophore hypothesis used here was presented as a useful basis for the development of anticancer compounds.
Keywords: Pharmacophore, 3c0z, HDAC7, QSAR
Histone deacetylases (HDACs) are known as a class of enzymes that regulate the elimination of acetyl groups from lysine residues of histones, thus playing an important regulatory role in epigenetics [1]. HDACs have been identified as a major player in tumorigenesis. Inhibition of HDAC function induces cell differentiation and suppresses cell proliferation, although it proved to be an effective strategy in the treatment of cancer [2]. Four classes of HDACs with eighteen subtypes are known to date and their presence in many cancers was reported [2]. Among them, HDAC7 has been particularly found in several types of cancers. Indeed, the role of HDAC7 was highlighted in cancers with a high level of HDAC7 protein observed in nine different cancers taken in pancreas of eleven humans [3]. Moreover, HDAC7 is over expressed in breast cancer stem cells and required to maintain cells [4]. HDAC7 also has a dual conflicting function in several types of cancer [5]. Recent reports indicated an ambivalent role of HDAC7 in cancers: for example, HDAC7 has an oncogenic function in childhood with acute lymphoblastic leukemia and pancreatic cancer [3] but at the same time, acts as a tumor suppressor in acute lymphoblastic leukemia B and Burkitt's lymphoma [6]. In addition, the high expression of HDAC7 was found to be related to a prognosis in lung cancer [7]. Clinical studies showed a correlation between HDAC7 inhibition and cancer treatment as the inhibition effects of HDAC7 lead to the death of cancer cells [8]. However, like any medicine, anticancer used substances often present troublesome effects for patients [9]. Since then, many drugs have been synthesized to target HDAC in order to induce cancer cells destruction. Despite recent advances and findings, the development of HDAC inhibitors as anticancer drug is still in its infancy. The main barrier remains the satisfactory balance between physicochemical and pharmacological and selectivity properties related to the candidate compounds. Compounds which display a promising anticancer activity with reduced side effects and necessary safety remain to be discovered. Pharmacophore model represents the 3D arrangements of the structural or chemical characteristics of a drug that may be essential to interact with enzyme to achieve an ideal binding. It should provide useful knowledge for the development of new potentially active candidates targeting HDAC7 in the treatment of cancer. Based upon the potential inhibitory properties of some compounds which were found to be active against HDAC7 [10], this study aims to design a 3D pharmacophore leading to develop new anticancer compounds with improved inhibition activity.
2.1 Selection of molecules and enzyme
The structure of the enzyme HDAC7 was downloaded from the PDB database (code: 3c0z) and then was purified according to the protocol available in the Discovery Studio 2.5 program [11]. A series of ten compounds recognized as HDAC7 inhibitors were selected [10]. The inhibitory concentrations of these compounds are given in Table 1. Eight (8) of them were employed for the training set and the two others, for the validation set.
Table 1. 2D-structures and biological activity values of selected compounds
The Discovery Studio 2.5 software was employed to generate the molecular modeling. Discovery Studio (DS) is a software suite for the simulation of small molecules and macromolecules. It is used as protein-modeling program in which the program tools allow visualizing, modifying, and simulating protein structures automatically, and also in accurate manner. The quality of DS offers enormous possibilities for the pharmacophore model starting from the ligand and crystallographic structures. The compounds are constructed and minimized by the CHARMM force field using Discovery.
2.2 Modeling of the pharmacophore
The ligands used in the training set were evaluated with the respect to their minimized conformations and subjected to the generation of pharmacophore (PH4) hypotheses. The characteristics of the pharmacophore were retained according to the compounds structures for generating pharmacophores based on the common characteristics displayed by the studied molecules. Discovery Studio is used to generate different models of pharmacophores, which are thereafter submitted to various validation criteria. Significant models are selected and validated based on excellent agreement between calculated and experimental binding constants, high correlation coefficient (R2), the lowest total cost and RMSD values [12]. The adequate alignment of the characteristic pharmacophores of respective ligands with those of the generated model is used to identify the promising leader pharmacophore model. Two theoretical cost calculations are critical to the success of the pharmacophore hypothesis. The "fixed cost" (the ideal cost) corresponds to the simplest model that fits perfectly to all the data, and the "null cost" (the cost without correlation) represents the pharmacophore cost without features. The difference between fixed cost and null cost constitutes a criterion too for the best model [13] [14]. The final model will be validated by a set of tested molecules. RMSD reflects the quality of the correlation between estimate and actual activity data. The generated pharmacophore model should be able to accurately describe activity of the molecules and identify the active compound from the database.
Among other important criteria in the field of drug design for chemists, the pharmacophore could be defined as the fundamental element. In this work, the pharmacophores hypotheses were elaborated following the docking QSAR model based on the Gibbs free energies of enzyme-ligand binding. Ten pharmacophore models with significant statistical parameters were generated. Table 2 presents all the hypotheses generated using Discovery Studio software.
Table 2. Statistical parameters for pharmacophores models of HDAC7 inhibitors
|
Hypothesis |
RMSD |
R2 |
Total cost |
aCost difference |
|
HYPO 1 |
1.1851 |
0.9850 |
34.1883 |
165.98 |
|
HYPO 2 |
1.5197 |
0.9749 |
36.6311 |
163.53 |
|
HYPO 3 |
1.5080 |
0.9753 |
36.6973 |
163.47 |
|
HYPO 4 |
1.5734 |
0.9730 |
37.2936 |
162.87 |
|
HYPO 5 |
1.6303 |
0.9710 |
38.1223 |
162.04 |
|
HYPO 6 |
1.6569 |
0.9700 |
38.1643 |
162.00 |
|
HYPO 7 |
1.6226 |
0.9713 |
38.4670 |
161.70 |
|
HYPO 8 |
1.6343 |
0.9709 |
39.8472 |
160.32 |
|
HYPO 9 |
1.6842 |
0.9693 |
40.1142 |
160.05 |
|
HYPO 10 |
1.6554 |
0.9702 |
40.2080 |
159.96 |
|
Fixed cost |
0.0000 |
1.0000 |
27.1584 |
173.01 |
|
Hypo Null |
6.8192 |
0.0000 |
200.1650 |
0.00 |
Fixed cost = 27.1584; Null cost = 200.1650; a Cost difference = null cost - total cost
Analysis of the values in Table 2 reveals interesting statistical characteristics for the ten generated pharmacophores hypotheses. Overall, the difference between the RMSD values of the hypotheses generated and that of the null hypothesis extends from 4.1350 (with HYPO 9) to 4.2341 (with HYPO 1) when that between these hypotheses and the fixed extends from 1.1851 (with HYPO 1) to 1.6892 (with HYPO 19). These pharmacophores models therefore lead to bind strongly to the active sites of HDAC7 enzyme as corroborated by the correlation coefficients which are all close to unity. Furthermore, the cost differences, all greater than 60 and the total costs, all less than 60 indicate a good binding establishment of the different molecule’s pharmacophores in the active sites of the enzyme. Consequently, the pharmacophores models defined according to the hypotheses constitute basic criteria that can be used for developing HDAC7 inhibitor compounds. However, the pharmacophore model which achieves optimal ligand-protein interaction is that for which the candidate inhibitor compounds best satisfy the steric and electrostatic characteristics required for a high probability of biological activity. Therefore, a screening was performed by superimposing the pharmacophores of the different ligands on the pharmacophores of each hypothesis generated in order to identify the hypothesis which achieves the best model of pharmacophores based on these ligands. On all the pharmacophores of the ligands explored in this work and the hypotheses of pharmacophores generated, the good superposition of ligand structures was observed with pharmacophores model HYPO 2. The main characteristics of HYPO 2 generated by Discovery Studio 2.5 based on the selected data are shown in Figure 1.
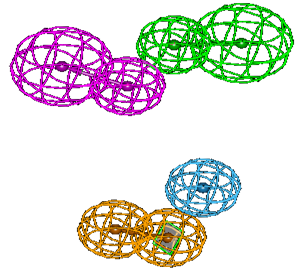
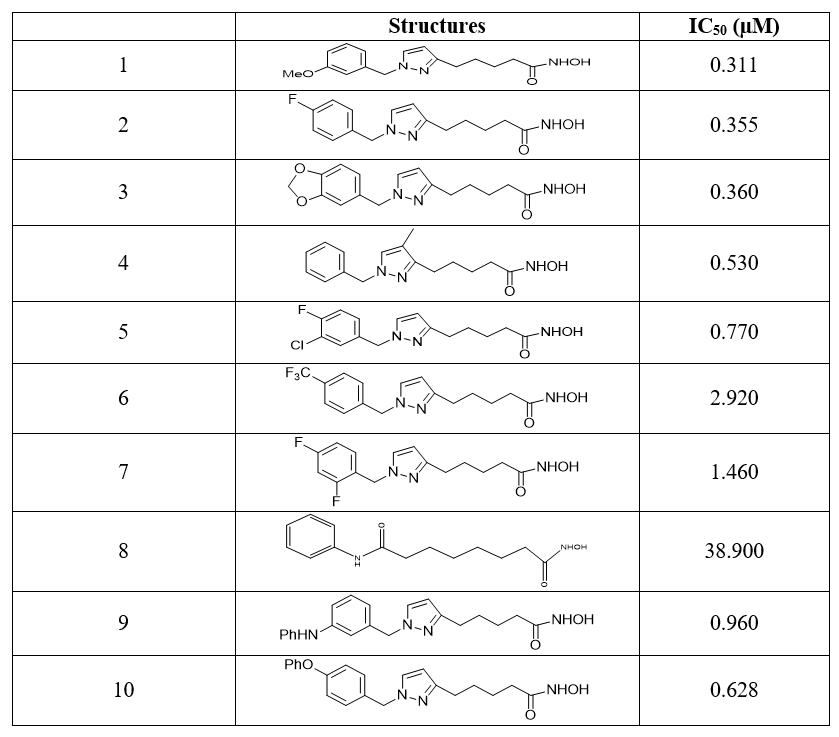
Figure 1. Main characteristics of the pharmacophore model HYPO 2.
Four main characteristics are identified for the pharmacophore model HYPO 2. These are the characteristics of hydrogen bond donor (HBD) in purple, of hydrogen bond acceptor (HBA) in green, aromatic rings (Ar) in orange and hydrophobicity (Hy) in blue. These characteristics are proved to be essential in the achievement of a better inhibition of enzyme HDAC7. The geometry parameters which define the spatial constraint arrangements of the main parts of the pharmacophore model HYPO 2 are shown in Figure 2 where (a) presents the distance constraints and (b) the angle constraints.
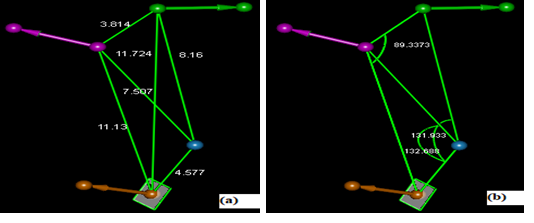
Figure 2: Geometry constraints in model HYPO 2: (a) distance (in Å); (b) angles (in º).
The pharmacophore parts related to hydrogen bond are separated to 3.814 Å when the parts requiring the presence of aromatic rings and hydrophobic groups are distant by 4.577 Å. The greatest distance (11.724 Å) is that which diametrically separates the parts of hydrogen bond acceptor and aromatic rings. The distance, on the other hand, which diametrically separates the point HBD from the point Hy is short (7.507 Å). Concerning the angles constraints, it is noticeable the characteristics points HBD, HBA and Ar practically describe a perpendicular plane (89.3373º). The geometry constraints in the HYPO 2 model reveal an arrangement that not only minimize the effects of polar and electrostatic interactions but also the likely competitions that could occur during interactions. For example, the distance between the points HBD and Ar precludes any competition for binding to a hydrogen bond acceptor. Indeed, aromatic rings are susceptible to hydrogen bonding. The results of the superposition of the ligands pharmacophores with those generated by the model HYPO 2 are given in Table 3. The used code “+” reflects the correct pairing of characteristic pharmacophores of ligand with those of model HYPO 2.
Table 3. Results of superimpositions of the pharmacophores of the ligands and of the model HYPO 2
|
No. |
Hy |
DLH |
ALH |
Ar |
|
1_25 |
+ |
+ |
+ |
+ |
|
2_17 |
+ |
+ |
+ |
+ |
|
3_28 |
+ |
+ |
+ |
+ |
|
4_31 |
+ |
+ |
+ |
+ |
|
5_27 |
+ |
+ |
+ |
+ |
|
6_15 |
+ |
+ |
+ |
+ |
|
7_30 |
+ |
+ |
+ |
+ |
|
8_SAHA |
+ |
+ |
+ |
+ |
|
9_23 |
+ |
+ |
+ |
+ |
|
10_19 |
+ |
+ |
+ |
+ |
Overall, the characteristic pharmacophores of the respective compounds well identify with those of the model. The pharmacophores of the studied ligands correspond well to the characteristic pharmacophores required to achieve a good ligand-protein interaction from the model HYPO 2. Observation of the superimposed structures, using Discovery Studio, made it possible to identify the ligand whose characteristic spheres of pharmacophores well correlated with those of the model HYPO 2. The best superposition was observed with the ligand 1_25 which, moreover, proved to be the most active compound of the series. Figure 3 shows the arrangements of pharmacophores in the ligand 1_25 according to the characteristics of hypothesis 2.
Figure 3. Characteristic arrangement of pharmacophores in the ligand 1_25
The arrangement of the pharmacophore characteristic spheres along ligand 1_25 molecular structure well corresponds to the molecule environment and to the expected properties related to the main hypotheses of the model HYPO 2. Aromatic characteristics are located around the benzene ring while hydrogen bond characteristics (HBD and HBA) involved the hydroxamic acids (-CONHOH) function. Nitrogen and oxygen atoms are of course likely to hydrogen bonding. The hydrophobic part is located on pyrazole ring. On the other hand, the pharmacophore model was used to estimate the biological activities of the ligands and the results were correlated with the experimental activities as shown in Figure 4. Two compounds were used for external validation and the values are reported in Table 5. This table presents the experimental (exp) inhibitory potentials and those estimated (est.) by the pharmacophore. The biological activity was expressed by the potential of inhibition concentration calculated following the relation:
pIC50exp=- log_10 (IC50 )
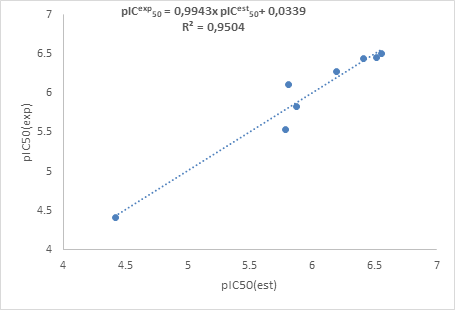
Figure 4. Correlation curve between experimental and estimated potentials
Table 5. Estimated and experimental values of the external validation set
| pIC50exp | pIC50ext | pIC50exp/pIC50ext | |
|
9_23 |
6.018 |
6.017 |
1.000 |
|
10_19 |
6.202 |
6.201 |
1.000 |
The analysis of the training set showed a 95.04% similarity between the experimental values and estimated values by the pharmacophore model. About the external validation set, we also observe a quasi-similarity between the experimental and estimated inhibitory potentials using the model of pharmacophore. The estimated biological activities from the model are close to those of the training set. The generated pharmacophore model of this work would be useful for the new HDAC7 inhibitors development to fight against cancer cell proliferation.
In this study, a pharmacophore model was designed using anticancer compounds. The work showed how the chemical characteristics of a set of compounds made it possible to generate hypotheses of pharmacophores from their biological activities. The best pharmacophore model did not only predict the compounds activities of the training set, but also compounds of the validation set. Compounds with different inhibitory activities were used to develop the pharmacophore model with high predictive power (95.04%) for the biological activity of HDAC7 inhibitors. Four distinct chemical characteristics were found and could be responsible HDAC7 inhibitors activity. These characteristics are hydrogen bond acceptor, hydrogen bond donor, aromatic ring and the hydrophobic character of the ligands. They have been classified as the essential characteristics for future drug candidates. The generated model was applied and then validated for two of the compounds. Both have an activity estimated such as the starting molecules and could therefore be used for further studies. Future drug candidates should therefore respect the characteristics generated by Hypothesis 2 in order to effectively target and inhibit HDAC7 in cancer cells.
O. Witt, H. E. Deubzer, T. Milde, and I. Oehme, "HDAC family: What are the cancer relevant targets?," Cancer Lett., vol. 277, no. 1, pp. 8-21, 2009. PMid:18824292
View Article PubMed/NCBIS. Yoon and G. H. Eom, "HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases," Chonnam Med. J., vol. 52, no. 1, pp. 1-12, 2016. PMid:26865995
View Article PubMed/NCBIM. Ouaïssi et al., "High Histone Deacetylase 7 (HDAC7) Expression Is Significantly Associated with Adenocarcinomas of the Pancreas," Ann. Surg. Oncol., vol. 15, no. 8, pp. 2318-2328, 2008. PMid:18506539
View Article PubMed/NCBIA. E. Witt et al., "Identification of a cancer stem cell-specific function for the histone deacetylases, HDAC1 and HDAC7, in breast and ovarian cancer," Oncogene, vol. 36, no. 12, pp. 1707-1720, 2017. PMid:27694895
View Article PubMed/NCBIM. Parra, "Class IIa HDACs - New insights into their functions in physiology and pathology," FEBS J., vol. 282, no. 9, pp. 1736-1744, 2015. PMid:25244360
View Article PubMed/NCBIB. Barneda-Zahonero et al., "The transcriptional repressor HDAC7 promotes apoptosis and c-Myc downregulation in particular types of leukemia and lymphoma," Cell Death Dis., vol. 6, no. 2, pp. 16-35, 2015. PMid:25675295
View Article PubMed/NCBIY. Lei et al., "Hdac7 promotes lung tumorigenesis by inhibiting Stat3 activation," Mol. Cancer, vol. 16, no. 1, pp. 1-13, 2017. PMid:29126425
View Article PubMed/NCBIM. Mottamal, S. Zheng, T. L. Huang, and G. Wang, "Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents.," Molecules, vol. 20, no. 3, pp. 3898-3941, Mar. 2015. PMid:25738536
View Article PubMed/NCBIT. Caruba, E. Jaccoulet, T. Caruba, and E. Jaccoulet, "Chimiothérapie anticancéreuse," Pharmacol. thérapeutiques, vol. 2, no. 1, pp. 57-80, 2015.
View ArticleYiwu Yao, Chenzhong Liao, Zheng Li, Zhen Wang, Qiao Sun, Chunping Liu and S. J. Yang Yang, Zhengchao Tu, "European Journal of Medicinal Chemistry Design , synthesis , and biological evaluation of 1 , 3-disubstituted- pyrazole derivatives as new class I and IIb histone deacetylase inhibitors," Eur. J. Med. Chem., vol. 86, pp. 639-652, 2014. PMid:25218912
View Article PubMed/NCBIA. Schuetz et al., "Human HDAC7 harbors a class IIa histone deacetylase-specific zinc binding motif and cryptic deacetylase activity.," J.Biol.Chem., vol. 283, pp. 11355-11363, 2008. PMid:18285338
View Article PubMed/NCBIA. N'Guessan, E. Megnassan1, Y. T. N. , Nahossé Ziao , Vladimir Frecer, and and S. Miertus, "Journal of Drug Design and Development," J. Drug Des. Dev., vol. 1, no. 1, pp. 11-28, 2017.
N. Kandakatla and G. Ramakrishnan, "Ligand Based Pharmacophore Modeling and Virtual Screening Studies to Design Novel HDAC2 Inhibitors," Adv. Bioinformatics, vol. 14, no. 1, pp. 1-11, 2014. PMid:25525429 4265523
View Article PubMed/NCBIN. Suresh and N. S. Vasanthi, "Pharmacophore Modeling and Virtual Screening Studies to Design Potential Protein Tyrosine Phosphatase 1B Inhibitors as New Leads," J. Proteomics Bioinform., vol. 03, no. 01, pp. 020-028, 2010.
View Article