Melalie Keïta
E-mail: keitamelalie@yahoo.fr
Tel.: +2250707829340
© 2019 Sift Desk Journals. All Rights Reserved
VOLUME: 5 ISSUE: 2
Page No: 606-630
Melalie Keïta
E-mail: keitamelalie@yahoo.fr
Tel.: +2250707829340
Yéo Yaya 1, Melalie Keïta 1,*, Bibila Mayaya Bisseyou Yvon 2, Akori Elvice Esmel 1, Brice Dali 1, Hermann N’Guessan 1
[1] Laboratoire de Physique Fondamentale et Appliquée (LPFA), University of Abobo Adjamé (now Nangui Abrogoua), Abidjan, Côte d’Ivoire; yeoyaya532@gmail.com (Y.Y.), keitamelalie@yahoo.fr (M.K.), elvicee@yahoo.fr (A.E.E.), dalibrice08@gmail.com (D.B.), nerhminos@yahoo.fr (H.N.)
[2] Laboratoire de Cristallographie – Physique Moléculaire, University of Cocody (now Felix Houphouët-Boigny), Abidjan, Côte d’Ivoire; bibilamayayabisseyou@gmail.com
Melalie Keita, Yéo Yaya, Bibila Mayaya Bisseyou Yvon , Akori Elvice Esmel, Brice Dali, Hermann N’Guessan, Molecular and Thermodynamic Modeling of the Protein-Ligand Interaction. Application to Computer-Assisted Design of Anti-Competitive Inhibitors of Human Histone Deacetylase 2 (HDAC2). (2021) Journal of Computational Chemistry & Molecular Modeling 5(2) p:605-630
Background: Histone deacetylases (HDACs) are promising drug targets for a variety of therapeutic applications. Here we report in silico design and evaluation of novel amine-based hydroxamic acid derivatives (DAHAs), HDAC2 inhibitors with favorable predicted pharmacokinetic profiles.
Methods: By using in situ modifications of the crystal structure of suberoylanilide hydroxamic acid SAHA-HDAC2 complex (PDB entry 4LXZ), 3D models of HDAC2-DAHAx complexes were prepared for a training set of 18 DAHAs with experimentally determined inhibitory potencies (half-maximal inhibitory concentrations IC50exp In the search for active conformations of the DAHA1-18, a linear QSAR model was prepared, which correlated computed gas-phase enthalpies of formation (∆∆HMM) of HDAC2-DAHAx complexes with the IC50exp. Further, taking into account the solvent effect and entropy changes upon ligand, binding resulted in a superior QSAR model correlating computed complexation Gibbs free energies (∆∆Gcom). The successive pharmacophore model (PH4) generated from the active conformations of DAHAs served as a virtual screening tool of novel analogs included in a virtual combinatorial library (VCL) of compounds containing hydroxamic acid scaffolds. The PH4 model to identify new DAHA analogs screened the VCL filtered by Lipinski’s rule-of-five.
Results: Gas-phase QSAR model: -log10(IC50exp) = pIC50exp -0.2870 x ∆∆HMM +6.5764, R2= 0.83, superior aqueous phase QSAR model: pIC50exp =-0.4005 x ∆∆Gcom + 6.4402, R2=0.93 and PH4 pharmacophore model : pIC50exp = 1.0107 x pIC50pre - 0.0607, R2 = 0.92. The VCL of more than 198 thousand DAHAs was filtered down to 150,713 analogs Lipinski’s rule. The PH4 screening retained 110 new and potent DAHAs with predicted inhibitory potencies pIC50pre up to 520-fold lower than .that of DAHA1 (IC50exp =260nM). Predicted pharmacokinetic profiles of the new analogs were compared to current per oral anti-cancer drugs.
Conclusions: This computational approach, which combines molecular modeling, pharmacophore model, analysis of HDAC2-DAHAs interaction energies, in silico screening of VCL of DAHAs, and ADME properties resulted in a set of proposed new HDAC2 inhibitors.
Keywords: histone deacetylase 2; amine-based hydroxamic acid derivatives; molecular modeling; QSAR models; pharmacophore; combinatorial library; in silico screening; ADME properties prediction.
Cancer is a generic term for any disease in which certain body cells mutate and divide uncontrolled. It is a set of undifferentiated cells that escape the control of the body, multiply indefinitely, invade nearby tissues by destroying them, and spread in the body during a process called metastasis. If the cancerous cells are not eliminated, the course of the disease will lead more or less quickly to death. These abnormal cells, therefore, represent a health hazard due to their ability to invade other healthy tissues. Cancer is a major public health problem worldwide. Indeed, it is the second leading cause of death in the world of about ten million deaths per year (nearly one in six deaths is due to cancer worldwide) [1]. There are several types of cancers, which are determined according to histology that is, depending on the nature of the tissue in which they develop. Thus, we distinguish carcinomas, sarcomas, and hematopoietic cancers. For decades, studies on the origin of cancer have shown that genetic alteration (mutations, amplification or loss of chromosomal material, recurrent translocations) was the cause. Nowadays, with the explosion of knowledge in molecular biology, it has become clear that the initiation and progression of cancer can be epigenetic.
Epigenetic deregulations encompass several modes of control that include DNA methylation, post-translational modifications (PTM) affecting the N-terminal part of histones. Post-translational changes include acetylation, methylation, phosphorylation, ubiquitination and sumoylation [2]. Specific enzymes that target certain specific amino acids of the N-terminal tail of histones catalyze them [3]. The acetylation level of histones is dependent on the antagonistic action of two families of enzymes: histone acetyltransferases (HATs) and histone deacetylases (HDACs) (Figure 1) [4].
HDACs play a significant role in the epigenetic regulation process of gene transcription and expression through their effects on the chromatin compaction state. Their inappropriate recruitment contributes to the development of cancers and their inhibition leads to genetic reprogramming in cancer cells.
Recent studies have shown that HDACs are promising therapeutic targets because of their potential to reverse the aberrant epigenetic states associated with carcinogenesis [5]. In humans, 18 HDAC enzymes have been identified and classified into four groups according to their homology with yeast HDACs [5]. Classes I, II, and IV require a zinc molecule, as a cofactor, in their active site whilst class III HDACs with similar structure homologous to the yeast Sir2 protein need NAD+ as a cofactor. Class I HDACs include HDACs 1, 2, 3, and 8 whose structures are similar to yeast Rpd3. Class II HDACs can be further subdivided into class IIa (HDACs 4, 5, 7, and 9) and class IIb (HDACs 6 and 10). As for HDAC11, it is the sole component of class IV.
The Food Drug Administration (FDA) Currently approves five anticancer agents with HDAC-mediated mechanisms of action: vorinostat, belinostat, romidepsin, tucidinostat, and panobinostat [6]. The first clinically successful HDAC inhibitor (HDACi) is the suberoylanilide hydroxamic acid (SAHA), also known as vorinostat (Zolinza®) [6]. It is an orally available pan-HDAC inhibitor for the treatment of cutaneous T-cell lymphoma (CTCL) [6]. Other agents of the hydroxamate class are belinostat (Beleodaq®), an intravenous pan-HDAC inhibitor for peripheral T-cell lymphoma (PTCL), and panobinostat (Farydak®), another orally active pan-HDAC inhibitor [6]. The non-hydroxamic benzamides class includes tucidinostat (Epidaza®), also referred to as chidamide, which is an orally active class 1 and 2-specific agent that was approved by China's National Medical Products Administration in 2014 for use in PTCL and in 2019 for use in postmenopausal advanced breast cancer patients in combination with exemestane, a steroidal aromatase inhibitor [6]. Romidepsin (Istodax®), an intravenous cyclic depsipeptide, is also specific to classes I and II and was approved by the FDA in 2009 for CTCL and in 2011 for PTCL [6].
HDAC2 in particular acts as a transcriptional repressor by deacetylation of lysine residues present at the N-terminal tail of histone proteins (H2A, H2B, H3, and H4). Thus, the study of its inhibition offers promising prospects for the development of future anti-tumor agents and cancer treatments [7, 8]. The HDAC2 active site pocket could be subdivided into four sub-pockets: the first sub-pocket, which is the catalytic center, contains the Zn2+ ion [9] and the residues His145, His146, Asp181, His183, Asp269, and Tyr308 [10, 11]. The second sub-pocket, which leads from the surface to the catalytic center, is connected to Gly154, Phe155, His183, Phe210, and Leu276 [9, 11]. The third sub-pocket located on the surface, looking towards the solvent, is connected to the polar residues Glu103, Asp104, Arg275, and non-polar residues Gly32, His33, Pro34 Met35 [10, 12]. The fourth sub-pocket called the "foot pocket" containing mainly water molecules is connected to Tyr29, Met35, Phe114, and Leu144 [9].
In this work, we design new analogs of amine-based hydroxamic acid derivatives (DAHA) from a series of 18 known DAHAs with specific experimental inhibition activities (IC50exp), which have been used as a training set (TS) of HDACi [12]. Amines are highly bioavailable and are promising candidates for further development for a variety of therapeutic applications [6]. Compounds in this series display excellent therapeutic capacity for a variety of anti-cancer applications.
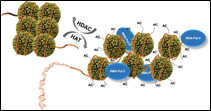
Figure 1. Regulation of gene expression and repression by histone acetylase (HAT) and histone deacetylase (HDAC) [4].
The workflow describing the steps of the entire process of virtual design of novel DAHA analogs is presented in scheme 1.
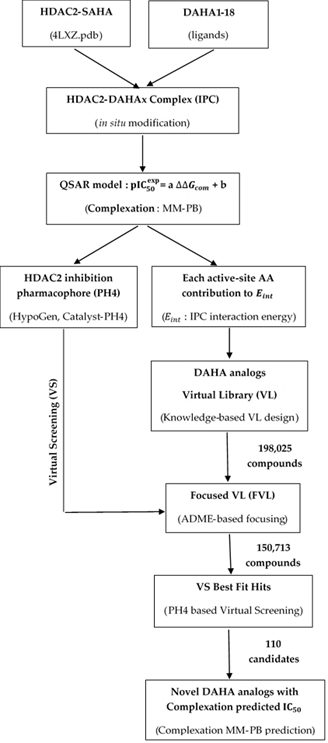
Scheme 1. Workflow describing the multistep approch to virtual design novel DAHA analogs with higher predicted potency against HDAC2.
2.1. Training and Validation Sets
Chemical structures and biological activities (IC50exp) of training and validation sets of amine-based hydroxamic acid derivatives inhibitors of human HDAC2 used in this study were taken from the literature [12]. The potencies of these compounds cover a sufficiently broad range of half-maximal inhibitory concentrations (260 ≤ IC50exp ≤ 15000nM) to allow the construction of a QSAR model. The training set (TS) containing 18 DAHA inhibitors and the validation set (VS) including 3 DAHAs were taken from the ref. [12].
2.2. Model Building
Three-dimensional (3D) molecular models of enzyme–inhibitor (E-I) complexes HDAC2-DAHAx, free enzyme HDAC2, and free inhibitors DAHA were prepared from the high-resolution (1.85 Å) crystal structure of a reference complex containing the compound SAHA inhibitor (PDB entry code: 4LXZ [11]) using the Insight-II molecular modeling program [13].
The structures of HDAC2 and the E-I complexes were considered to be at a pH of 7 with neutral N- and C-terminal residues and all protonizable and ionizable residues charged. No crystallographic water molecules are included in the model. The inhibitors were built into the reference structure 4LXZ [11] by in situ replacing of derivatized groups in the molecular scaffold of the template inhibitor SAHA. An exhaustive conformational search over all rotatable bonds of the replacing function groups coupled with a careful gradual energy-minimization of the modified inhibitor and active site residues of the HDAC2 located in the vicinity of the inhibitor (within 5 Å distance) was employed to identify low-energy bound conformations of the modified inhibitor. The resulting low-energy structures of the E-I complexes were carefully refined by minimization of the whole complex. This procedure has been successfully used for model building of viral, bacterial, and protozoal enzyme–inhibitor complexes and design of peptidomimetic, hydroxynaphthoic, thymidine, triclosan, pyrrolidine carboxamide, nitriles, and chalcone-based inhibitors [14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25].
2.3. Molecular Mechanics
Modeling of inhibitors, HDAC2, and E-I complexes were conducted by molecular mechanics using CFF91 force field [26] as described earlier [14].
2.4. Conformational Search
Free inhibitor conformations were derived from their bound conformations in the E-I complexes by gradual relaxation to the nearest local energy minimum as described earlier [14].
2.5. Solvation Gibbs Free Energies
The electrostatic component of solvation Gibbs free energy (GFE) that includes also the effects of ionic strength via solving nonlinear Poisson–Boltzmann equation [27, 28] was computed by the Delphi module in Discovery Studio [29] as described earlier [14].
2.6. Calculation of Binding Affinity and QSAR Model
The calculation of binding affinity expressed as complexation GFE has been described fully earlier [14].
2.7. Interaction Energy
The calculation of MM interaction energy between enzyme residues and the inhibitor CFF91 force field [26] was performed as described earlier [14].
2.8. Pharmacophore Generation
Bound conformations of inhibitors taken from the models of E-I complexes were used for constructing of 3D-QSAR pharmacophore (PH4) by using Catalyst HypoGen algorithm [30] implemented in Discovery Studio [29] as described earlier [14].
2.9. ADME Properties
The QikProp program [31] computed the pharmacokinetics profile of DAHAs as described earlier [14].
2.10. Virtual Library Generation
The virtual library generation was performed as described earlier [14].
2.11. ADME-Based Library Searching
The drug-likeness selection criterion served to focus the initial virtual library as described earlier [14].
2.12. Pharmacophore-Based Library Searching
The pharmacophore model (PH4) described in Section 2.8 and derived from the bound conformations of DAHAs at the active site of HDAC2 served as a library searching tool as described earlier [14].
2.13. Inhibitory Potency Prediction
The conformer with the best mapping on the PH4 pharmacophore in each cluster of the focused library subset was used for ∆∆Gcom = calculation and IC50pre estimation (virtual screening) by the complexation QSAR model as described earlier [14].
3.1. Training and Validation Sets
A training set of 18 DAHAs and a validation set of 3 DAHAs (table 1) were selected from a homogeneous series of class I HDACs inhibitors for which experimentally determined inhibitory activities were available from a single laboratory [12]. The whole series was obtained by variations at two positions R1 and R2 of the amino group as shown in Table 1. The experimental half-maximal inhibitory concentrations (260 ≤ IC50exp ≤ 15000nM)[12] cover a sufficiently wide concentration range for building of a reliable QSAR model.
Table 1. Training Set (TS) and Validation Set (VS) of DAHA inhibitors [12] of human HDAC2 used in the preparation of quantitative structure-activity relationships (QSAR) model of inhibitor binding.

3.2. QSAR Model
3.2.1. One Descriptor QSAR Models
Each of the 18 training sets (TS) and 3 validation sets (VS) HDAC2-DAHAx complexes (Table 1), was prepared by in situ modification of the refined template crystal structure (PDB code 4LXZ [11]) of the complex HDAC2-SAHA as described in the Methods section. Further, the relative Gibbs free energy of the HDAC2-DAHAx complexes formation(∆∆Gcom) was computed for each of the 21 optimized enzyme-inhibitor complexes. Table 2 lists computed values of ∆∆Gcom and its components for the TS and VS of DAHAs [12]. The QSAR model explained variation in the DAHAs experimental inhibitory potencies (pIC50exp = -log10(IC50exp)[12]) by correlating it with computed GFE ∆∆Gcom through linear regression (Equation (B), Table 3). In addition, a significant correlation obtained in this QSAR relationship permitted the active bound conformation of the DAHAs at the HDAC2 binding site and enabled the definition of the PH4 pharmacophore. In search for a better insight into the binding affinity of DAHAs towards HDAC2, we have analyzed the enthalpy of complexation in gas-phase ∆∆HMM by correlating it with the pIC50exp. The validity of this linear correlation (for statistical data of the regression see Table 3, Equation (A)) allowed assessment of the significance of inhibitor-enzyme interactions (∆∆HMM) when solvent effect and loss of entropy of the inhibitor upon binding to the enzyme were neglected. This correlation explained about 83% of the pIC50exp data variation and underlined the role of the enthalpic contribution to the binding affinity of the ligand. Similarly, the more advanced descriptors, namely the GFE of the HDAC2-DAHAx complex formation including all components: ∆∆HMM, ∆∆TSvib and ∆∆Gsol , have been assessed (for statistical data see Table 3, Equation (B)). Relatively high values of the regression coefficient R2, leave-one-out cross-validated regression coefficient R2xv and Fischer F-test of the correlation suggest a strong relationship between the 3D model of inhibitor binding and the observed inhibitory potencies of the DAHAs [12]. Therefore, structural information derived from the 3D models of HDAC2-DAHAx complexes can be expected to lead to the reliable prediction of HDAC2 inhibitory potencies for new DAHAs analogs based on the QSAR model B, Table 3.
Table 2. Gibbs free energy (binding affinity) and its components for the training set of human HDAC2 inhibitors DAHA1-18 and validation set inhibitors DAHA19-21 [12].
|
Training Set a |
Mwb |
∆∆HMM c |
∆∆Gsold |
∆∆TSvibe |
∆∆Gcomf |
IC50exp g |
|
[g·mol−1] |
[kcal·mol−1] |
[kcal·mol−1] |
[kcal·mol−1] |
[kcal·mol−1] |
[nM] |
|
|
DAHA1 |
290 |
0.0 |
0.0 |
0.0 |
0.0 |
260 |
|
DAHA2 |
300 |
1.0 |
-1.0 |
-0.3 |
0.3 |
320 |
|
DAHA3 |
330 |
1.1 |
-1.4 |
-0.9 |
0.6 |
690 |
|
DAHA4 |
280 |
2.3 |
-1.8 |
-0.4 |
0.9 |
790 |
|
DAHA5 |
290 |
2.6 |
-1.0 |
1.1 |
0.5 |
840 |
|
DAHA6 |
270 |
3.5 |
-1.4 |
0.8 |
1.3 |
1 500 |
|
DAHA7 |
310 |
2.1 |
-1.3 |
-1.4 |
2.2 |
1 800 |
|
DAHA8 |
340 |
2.6 |
-2.5 |
-2.3 |
2.4 |
2 700 |
|
DAHA9 |
300 |
4.4 |
-1.5 |
0.6 |
2.3 |
2 900 |
|
DAHA10 |
500 |
3.5 |
0.6 |
1.6 |
2.5 |
3 000 |
|
DAHA11 |
250 |
4.6 |
-1.0 |
1.5 |
2.1 |
3 100 |
|
DAHA12 |
250 |
4.1 |
-1.7 |
-0.5 |
2.9 |
3 200 |
|
DAHA13 |
420 |
4.4 |
0.6 |
2.5 |
2.5 |
3 700 |
|
DAHA14 |
340 |
3.3 |
-1.7 |
-1.2 |
2.8 |
4 000 |
|
DAHA15 |
440 |
3.5 |
-0.6 |
0.7 |
2.2 |
4 200 |
|
DAHA16 |
430 |
3.4 |
-0.8 |
0.4 |
2.2 |
4 200 |
|
DAHA17 |
380 |
5.0 |
-4.9 |
-3.6 |
3.7 |
10 000 |
|
DAHA18 |
410 |
5.8 |
1.8 |
3.7 |
3.9 |
15 000 |
|
Validation Set a |
Mw b |
∆∆HMM c |
∆∆Gsol d |
∆∆TSvib e |
∆∆Gcom f |
pIC50pre/pIC50exp h |
|
[g·mol−1] |
[kcal·mol−1] |
[kcal·mol−1] |
[kcal·mol−1] |
[kcal·mol−1] |
||
|
DAHA19 |
330 |
2.1 |
-0.9 |
0.0 |
1.1 |
0.97 |
|
DAHA20 |
460 |
2.4 |
-0.2 |
1.3 |
0.9 |
1.02 |
|
DAHA21 |
490 |
3.2 |
-2.1 |
-1.8 |
2.9 |
0.97 |
a for the chemical structures of the training set of inhibitors see Table 1; b Mw is the molar mass of inhibitors; c ∆∆HMM is the relative enthalpic contribution to the GFE change related to E-I complex formation derived by MM; ∆∆HMM = [EMM{E:Ix}- EMM {Ix}]-[EMM{E:Iref}- EMM {Iref}], Iref is the reference inhibitor DAHA1; d ∆∆Gsol is the relative solvent effect contribution to the GFE change of E-I complex formation: ∆∆Gsol= [Gsol{E:Ix}- Gsol{Ix}]-[Gsol{E:Iref}- Gsol{Iref}];e ∆∆TSvib is the relative entropic contribution of inhibitor Ix to the GFE of E-Ix complex formation: ∆∆TSvib= [TSvibl{Ix}E- TSvib{Ix}]-[TSvib{Iref}E- TSvib{Iref}]; f ∆∆Gcom is the overall relative GFE change of E-Ix complex formation: ∆∆Gcom = ∆∆HMM + ∆∆Gsol - ∆∆TSvib; g IC50exp is the experimental half-maximal inhibition concentration of human HDAC2 obtained from ref. [12]; h ratio of predicted and experimental half-maximal inhibition concentrations pIC50pre/pIC50exp (pIC50pre =-log10 IC50pre) was predicted from computed ∆∆Gcom using the regression equation for human HDAC2 shown in Table 3, (B).
Table 3. Analysis of computed binding affinities ∆∆Gcom, its enthalpic component, and experimental half-maximal inhibitory concentrations pIC50exp = -log10 (IC50exp) of DAHAs towards HDAC2 [12].
|
Statistical Data of Linear Regression |
(A) |
(B) |
|
pIC50exp= − 0.2870 × + 6.5764 (A) |
- |
- |
|
pIC50exp= − 0.4005 × + 6.4402 (B) |
- |
- |
|
Number of compounds n |
18 |
18 |
|
Squared correlation coefficient of regression R2 |
0.83 |
0.93 |
|
LOO cross-validated squared correlation coefficient R2xv |
0.82 |
0.93 |
|
Standard error of regression σ |
0.20 |
0.13 |
|
Statistical significance of regression, Fischer F-test |
78.98 |
217.76 |
|
Level of statistical significance α |
>95% |
>95% |
|
Range of activities IC50exp[nM] |
260-15 000 |
|
The statistical data confirmed the validity of the correlation Equations (A) and (B) plotted on Figure 2. The ratio pIC50pre/pIC50exp ≅ 1(the pIC50pre values were estimated using correlation Equation (B), Table 3) calculated for the validation set DAHA19-21 documents the substantial predictive power of the complexation QSAR model from Table 2. Thus, the regression Equation (B) (Table 3) and computed ∆∆Gcom GFE can be used for the prediction of inhibitory potencies IC50pre against HDAC2 for novel DAHA analogs, provided that they share the same binding mode as the training set DAHA1-18.
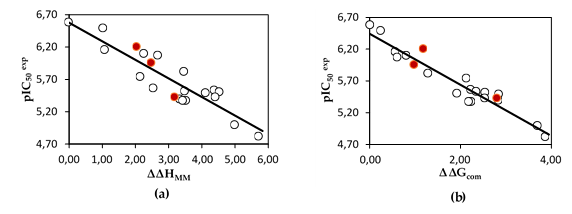
Figure 2. (a) Plot of correlation equation between pIC50exp and relative enthalpic contribution to the GFE ∆∆HMM; (b) Plot for relative complexation GFE ∆∆Gcom of the training set of DAHAs, all in kcal·mol−1. Validation set data is shown in red color.
3.2.2. Binding Mode of DAHAs
The structural analysis of the interactions of the HDAC2-DAHA1-16 complexes corroborates the experimental inhibitory activities obtained by Pavel et al. [12]. The key interactions involved in HDAC2-DAHAx complexes justifying their affinity are the number of hydrogen bonds (HBs), van der Waals (vdW), hydrophobic contacts, etc. FIG. 3 shows the binding mode of the most active ligand of the test set (DAHA1) as well as that of DAHA16, one of the least active. It reveals a better aptitude of the inhibitor DAHA1 in the binding pocket of HDAC2.
Indeed, the ZBG of DAHA1 makes several H bonds with the residues His145, His146, His183, Asp181, and Tyr308 (figure 3. a, b). The linker is well housed in the hydrophobic tunnel formed by the side chains of residues Gly154, Phe155, His183, Phe210, and Leu216. The DAHA1 SBG with the 1H -indol-2-yl substituent at the R1 position (fused bicyclic part) and the hydrogen at the R2 position (Table 1) shows two Pi-Cation interactions with Arg275 and one H bond interaction with Asp104 (Figure 3. a, b).
In the case of the DAHA16 inhibitor, the substitution of hydrogen by a fairly large fragment at position R2 resulted in the loss of two H bonds: one between ZBG and Asp181 and the other between SBG and Asp104 (Figure 3, d). In addition, the two aforementioned Pi-Cation interactions for DAHA1 were lost and replaced by a Pi-Donor Hydrogen bond interaction between the substituent at the R1 position of the DAHA16 scaffold and the Arg275 residue. This information reveals that DAHA1 exhibits more additional interactions with active site residues, which describes its binding potential to inhibit HDAC2 activity.
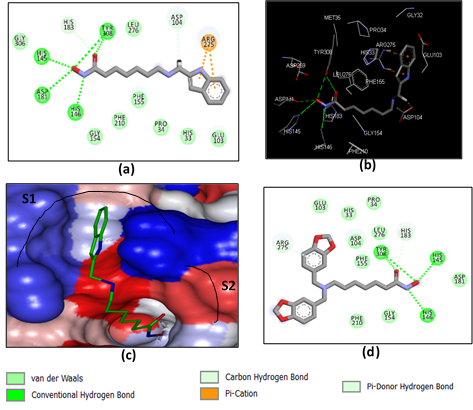
Figure 3. (a) 2D schematic interaction diagram of the most potent inhibitor DAHA1 [12] at the active site of human HDAC2; (b) 3D structure of the HDAC2 active site with bound inhibitor DAHA1; (c) Connolly surface of the HDAC2 active site for DAHA1. Surface coloring legend: red = hydrophobic, blue = hydrophilic, and white = intermediate; (d) 2D schematic interaction diagram of the inhibitor DAHA16 [12] at the active site of human HDAC2.
3.3. Interaction Energy
Other key structural information was provided by the Interaction Energy (IE, ∆Eint) diagram obtained for each training set inhibitor. IE breakdown to contributions from HDAC2 active site residues is helpful for the choice of relevant R1-groups and R2-groups, which could improve the binding affinity of DAHA analogs to the human HDAC2 and subsequently enhance the inhibitory potency. A comparative analysis of computed IE for the training set DAHAs (Figure 4) divided into three classes (highest, moderate, and lowest activity) has been conducted to identify the residues for which the contribution to binding affinity could be increased. However, the comparative analysis showed about the same level of IE contributions of the residues of the active site around the ZBG residues (His145, His146, Asp181, and Tyr308) and around the linker (Gly154, Phe155, His183, Phe210, and Leu276) for all three classes of inhibitors, which seems normal to us since it is the same linker and the same ZBG that are used in all the training set inhibitors in these pockets. The difference would then come from the contribution of the site I residues (Gly32, His33, Pro34, Met35, Glu103, Asp104, and Arg275) which form the pocket where the R1-groups are housed (Figure 3, c). the variation observed in the activity of the different DAHAs is due to a concerted action of the HDAC2 active site residues. A combinatorial approach was adopted to novel DAHA analogs design and in silico screened a virtual library of 198,025 DAHA analogs with help of the PH4 pharmacophore of HDAC2 inhibition derived from the complexation QSAR model. The charged amino acid residues Glu103, Asp104 and Arg275 from the site I (S1) as well as the hydrophobic amino acid residue Leu276 from the site II (S2) (figure 3, c) could increase the potency of the novel DAHA analogs.
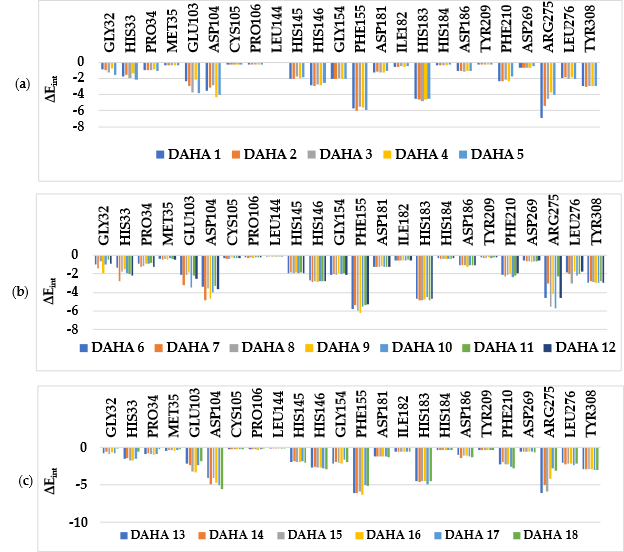
Figure 4. Molecular mechanics intermolecular interaction energy ∆Eint breakdown to residue contributions in [kcal.mol-1]: (a) the most active inhibitors DAHA1-5; (b) moderately active inhibitors DAHA6-12; (c) less active inhibitors DAHA7-18, Table 2 [12].
3.4. 3D-QSAR Pharmacophore Model
3.4.1. HDAC2 Active Site Pharmacophore
The Connolly surface generation protocol in Insight-II molecular modeling program [31] allows for mapping of hydrophobic and hydrophilic character of the active site of a protein. The surface of the active site of HDAC2 is both hydrophobic and hydrophilic (Figure 3, c).
3.4.2. Generation and Validation of 3D-QSAR Pharmacophore
HDAC2 inhibition 3D-QSAR pharmacophore was generated from the active conformation of 18 TS DAHA1-18 and evaluated by 3 VS DAHA19-21 covering a large range of experimental activity (260 – 15000 nM) spanning more than two orders of magnitude [12]. The generation process is divided into three main steps: (i) the constructive step, (ii) the subtractive step, and (iii) the optimization step [29] as described earlier [14]. During the constructive phase, DAHA1 and DAHA2 were retained as the lead (since their activities fulfilled the threshold criterion, IC50exp ≤ 2 x 260 nM) and used to generate the starting PH4 features. In the subtractive phase, compounds for which IC50exp > 260 x 103.5 nM = 822192 nM were considered inactive. Accordingly, none of the training set DAHAs was inactive and no starting PH4 features were removed. Finally, during the optimization phase, the score of the pharmacophoric hypotheses was improved. Hypotheses were scored according to errors in the estimated activity from regression and complexity via a simulated annealing approach. At the end of the optimization, the top-scoring 10 unique pharmacophore hypotheses were kept, all displaying three features. The cost values, correlation coefficients, root-mean-square deviation (RMSD) values, the pharmacophore features, and the max-fit value of the top 10 ranked hypotheses (Hypo1−Hypo10) are listed in Table 4. They are selected based on significant statistical parameters, such as high correlation coefficient, low total cost, and low RMSD.
Table 4. Parameters of 10 generated PH4 pharmacophoric hypotheses for HDAC2 inhibitors [12] after the CatScramble validation procedure (49 scrambled runs for each hypothesis at the selected level of confidence of 98%).
|
Hypothesis |
RMSD a | R2 b |
Total Costs c |
Costs Difference d |
Closest Random e |
|
Hypo1 |
1.349 |
0.96 |
60.2 |
169.3 |
80.51 |
|
Hypo2 |
2.217 |
0.88 |
92.0 |
137.5 |
91.15 |
|
Hypo3 |
2.283 |
0.87 |
94.3 |
135.2 |
104.03 |
|
Hypo4 |
2.506 |
0.84 |
106.7 |
122.8 |
108.76 |
|
Hypo5 |
2.581 |
0.83 |
110.5 |
119.0 |
109.57 |
|
Hypo6 |
2.597 |
0.83 |
111.1 |
118.4 |
111.19 |
|
Hypo7 |
2.686 |
0.82 |
112.2 |
117.3 |
115.42 |
|
Hypo8 |
2.667 |
0.82 |
112.6 |
116.9 |
116.57 |
|
Hypo9 |
2.677 |
0.82 |
112.9 |
116.6 |
117.81 |
|
Hypo10 |
2.658 |
0.82 |
114.2 |
115.3 |
119.43 |
a root-mean-square deviation; b squared correlation coefficient; c overall cost parameter of the PH4 pharmacophore; d cost difference between Null cost and hypothesis total cost; e lowest cost from 49 scrambled runs at a selected level of confidence of 98%. The Fixed Cost = 43.4 with RMSD = 0, the Null Cost = 229.5 with RMSD = 4.687 and the Configuration cost = 10.38.
The generated pharmacophore models were assessed for their reliability based on the calculated cost parameters ranging from 60.2 (Hypo1) to 114.2 (Hypo10). The relatively small gap between the highest and lowest cost parameter corresponds well with the homogeneity of the generated hypotheses and the consistency of the TS of DAHAx. For this PH4 model, the fixed cost (43.4) is lower than the null cost (229.5) by a difference ∆ = 186.1. This difference is a major quality indicator of the PH4 predictability (∆ > 70 corresponds to an excellent chance or a probability higher than 90% that the model represents a true correlation [29]). To be statistically significant, a hypothesis has to be as close as possible to the fixed cost and as far as possible from the null cost. For the set of 10 hypotheses, the difference ∆ ≥ 115.3, which attests to the high quality of the pharmacophore model. The standard indicators such as the RMSD between the hypotheses ranged from 1.349 to 2.658, and the squared correlation coefficient (R2) falls to an interval from 0.96 to 0.82. The first PH4 hypothesis with the closest cost (60.2) to the fixed one (43.4) and best RMSD and R2 was retained for further analysis. The statistical data for the set of hypotheses (costs, RMSD, R2) are listed in Table 4. The configuration cost (10.38 for all hypotheses) far below 17 confirms this pharmacophore as a reasonable one. The link between the 98% significance and the number 49 scrambled runs of each hypothesis is based on the formula S = [1-(1+X)/Y]x 100, with X the total number of hypotheses having a total cost lower than the original hypothesis (Hypo 1) and Y the total number of HypoGen runs (initial + random runs):X = 0 and Y = (1+49), hence 98% = {1-[(1+0)/(49+1)]}x100.
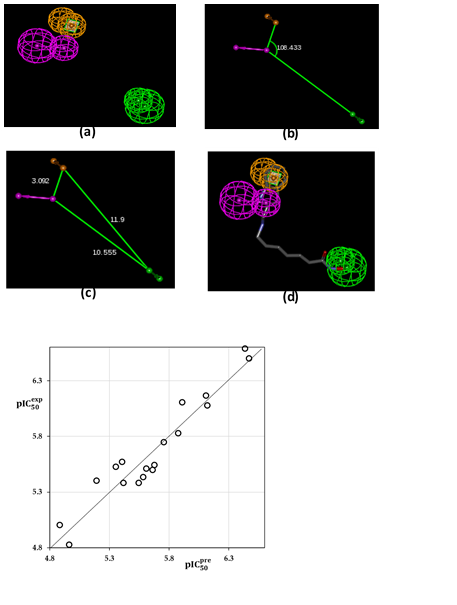
Figure 5. (a) features of the pharmacophore of HDAC2 inhibition; (b) angle between centers of pharmacophoric features; (c) distances between centers; (d) pharmacophore mapping with the most potent molecule DAHA1 (IC50exp = 260nM). The features are colored purple for Hydrogen Bond Donor (HBD), green for Hydrogen Bond Acceptor (HBA) and orange for Aromatic ring (Ar) ; (e) the correlation plot of experimental vs. predicted inhibitory activity (open circles correspond to TS).
The evaluation of Hypo 1 was performed first through Fischer’s randomization cross-validation test. The CatScramble program was used to randomize the experimental activities of the training set. At 98% confidence level, each of the 49 scramble runs created ten valid hypotheses, using the same features and parameters as in the generation of the original 10 pharmacophore hypotheses. Among them, the cost value of Hypo1 is the lowest compared with those of the 49 randomly generated hypotheses, as we can see in Table 4 where the lowest cost of the 49 random runs is listed for each original hypothesis, and none of them was as predictive as the original hypotheses generated shown in Table 4. Thus, there is a 98% probability that the best-selected hypothesis Hypo1 represents a pharmacophore model for inhibitory activity of DAHAs with a similar level of predictive power as the complexation QSAR model, which relies on the DAHAx active conformation from 3D structures of the HDAC2-DAHAx complexes and computed GFE of enzyme–inhibitor binding ∆∆Gcom. Another evaluation of Hypo 1 is the mapping of the best active training set DAHA1 (Figure 5) displaying the geometry of the Hypo1 pharmacophore of HDAC2 inhibition. The regression equation for pIC50pre vs pIC50exp estimated from Hypo1: pIC50exp = 1.0107 x pIC50pre - 0.0607(C)(n=18, R2 = 0.92, R2xv = 0.91, F -test = 177.40, σ =0.138, α >98%) is also plotted in Figure 5.
3.5. Virtual screening
In silico screening of a virtual library of ligands can lead to hits identification as it was shown in our previous works on inhibitor design [14, 15, 33, 34, 35].
3.5.1. Virtual Library
An initial virtual combinatorial library (VCL) was generated by substitutions at positions R1 and R2 (see Table 5) on the amine-based hydroxamic acid derivatives scaffold. During the VCL enumeration, the R-groups listed in Table 5 were attached to positions R1 and R2 of the DAHA scaffold to form a virtual combinatorial library of the size: R1 x R2 =445 x 445 = 198,025 DAHA analogs (Table 5). All analogs are matching the substitution pattern of the best inhibitor DAHA1. This DAHAs analogs library was generated from fragments (chemicals) listed in databases of available chemicals [36]. Nowadays, one of the criteria for the design of new anticancer drugs, for the target population, is their oral bioavailability. To design a more targeted library of reduced size and increased content of drug-like and orally bioavailable molecules, a set of filters and penalties were introduced, such as the Lipinski rule-of-five [37] facilitating the selection of a smaller number of suitable DAHAs that can be submitted to in silico screening. Thus, the initial library was reduced to 150,713 analogs, 76% of its initial size.
Table 5. R1- and R2-groups (fragments, building blocks, substituents) were used in the design of the initial diversity virtual combinatorial library of amine-based hydroxamic acid derivatives.
|
R-groupes a |
|||||
|
1 |
4-(5-fluoro-1H-pyrazol-1-yl)phenyl |
2 |
4-(4-fluoro-1H-pyrazol-1-yl)phenyl |
3 |
4-(3-fluoro-1H-pyrazol-1-yl)phenyl |
|
4 |
4-(3,4-difluoro-1H-pyrazol-1-yl)phenyl |
5 |
4-(3,4,5-trifluoro-1H-pyrazol-1-yl)phenyl |
6 |
4-(4,5-difluoro-1H-pyrazol-1-yl)phenyl |
|
7 |
4-(3,5-difluoro-1H-pyrazol-1-yl)phenyl |
8 |
4-(3-bromo-1H-pyrazol-1-yl)phenyl |
9 |
4-(4-bromo-1H-pyrazol-1-yl)phenyl |
|
10 |
4-(5-bromo-1H-pyrazol-1-yl)phenyl |
11 |
4-(4,5-dibromo-1H-pyrazol-1-yl)phenyl |
12 |
4-(3,4-dibromo-1H-pyrazol-1-yl)phenyl |
|
13 |
4-(3,5-dibromo-1H-pyrazol-1-yl)phenyl |
14 |
4-(3,4,5-tribromo-1H-pyrazol-1-yl)phenyl |
15 |
4-(5-mercapto-1H-pyrazol-1-yl)phenyl |
|
16 |
4-(4-mercapto-1H-pyrazol-1-yl)phenyl |
17 |
4-(3-mercapto-1H-pyrazol-1-yl)pheny |
18 |
4-(3,4-dimercapto-1H-pyrazol-1-yl)phenyl |
|
19 |
4-(4,5-dimercapto-1H-pyrazol-1-yl)phenyl |
20 |
4-(3,5-dimercapto-1H-pyrazol-1-yl)phenyl |
21 |
4-(3,4,5-trimercapto-1H-pyrazol-1-yl)phenyl |
|
22 |
4-(3-iodo-1H-pyrazol-1-yl)phenyl |
23 |
4-(4-iodo-1H-pyrazol-1-yl)phenyl |
24 |
4-(5-iodo-1H-pyrazol-1-yl)phenyl |
|
25 |
4-(4,5-diiodo-1H-pyrazol-1-yl)phenyl |
26 |
4-(3,4-diiodo-1H-pyrazol-1-yl)phenyl |
27 |
4-(3,4,5-triiodo-1H-pyrazol-1-yl)phenyl |
|
28 |
4-(3,5-diiodo-1H-pyrazol-1-yl)phenyl |
29 |
4-(3-chloro-1H-pyrazol-1-yl)phenyl |
30 |
4-(4-chloro-1H-pyrazol-1-yl)phenyl |
|
31 |
4-(5-chloro-1H-pyrazol-1-yl)phenyl |
32 |
4-(4,5-dichloro-1H-pyrazol-1-yl)phenyl |
33 |
4-(3,5-dichloro-1H-pyrazol-1-yl)phenyl |
|
34 |
4-(3,4-dichloro-1H-pyrazol-1-yl)phenyl |
35 |
4-(3,4,5-trichloro-1H-pyrazol-1-yl)phenyl |
36 |
4-(3-amino-1H-pyrazol-1-yl)phenyl |
|
37 |
4-(4-amino-1H-pyrazol-1-yl)phenyl |
38 |
4-(5-amino-1H-pyrazol-1-yl)phenyl |
39 |
4-(4,5-diamino-1H-pyrazol-1-yl)phenyl |
|
40 |
4-(3,5-diamino-1H-pyrazol-1-yl)phenyl |
41 |
4-(3,4-diamino-1H-pyrazol-1-yl)phenyl |
42 |
4-(3,4,5-triamino-1H-pyrazol-1-yl)phenyl |
|
43 |
4-(3-methyl-1H-pyrazol-1-yl)phenyl |
44 |
4-(4-methyl-1H-pyrazol-1-yl)phenyl |
45 |
4-(5-methyl-1H-pyrazol-1-yl)phenyl |
|
46 |
4-(4,5-dimethyl-1H-pyrazol-1-yl)phenyl |
47 |
4-(3,5-dimethyl-1H-pyrazol-1-yl)phenyl |
48 |
4-(3,4-dimethyl-1H-pyrazol-1-yl)phenyl |
|
49 |
4-(3,4,5-trimethyl-1H-pyrazol-1-yl)phenyl |
50 |
4-(5-ethyl-1H-pyrazol-1-yl)phenyl |
51 |
4-(4-ethyl-1H-pyrazol-1-yl)phenyl |
|
52 |
4-(5-ethyl-4-methyl-1H-pyrazol-1-yl)phenyl |
53 |
4-(5-ethyl-3,4-dimethyl-1H-pyrazol-1-yl)phenyl |
54 |
4-(5-(methylthio)-1H-pyrazol-1-yl)phenyl |
|
55 |
4-(4-mercapto-5-(methylthio)-1H-pyrazol-1-yl)phenyl |
56 |
4-(4,5-bis(methylthio)-1H-pyrazol-1-yl)phenyl |
57 |
4-(4-mercapto-3-methyl-5-(methylthio)-1H-pyrazol-1-yl)phenyl |
|
58 |
4-(5-(aminothio)-1H-pyrazol-1-yl)phenyl |
59 |
4-(4-(aminothio)-1H-pyrazol-1-yl)phenyl |
60 |
4-(4-(aminothio)-5-mercapto-1H-pyrazol-1-yl)phenyl |
|
61 |
4-(4,5-bis-(aminothio)-1H-pyrazol-1-yl)phenyl |
62 |
1,1’-biphenyl-4-yl |
63 |
4-(5H-tetrazol-5-yl)phenyl |
|
64 |
4-(1H-imidazol-1-yl)phenyl |
65 |
4-(1H-1,2,4-triazol-1-yl)phenyl |
66 |
4-(1H-tetrazol-1-yl)phenyl |
|
67 |
4-(thiophen-2-yl)phenyl |
68 |
4-(pyrazin-2-yl)phenyl |
69 |
4-(pyrimidin-2-yl)phenyl |
|
70 |
4-(pyridazin-3-yl)phenyl |
71 |
4-(piperazin-1-yl)phenyl |
72 |
3H-indol-2-yl |
|
73 |
7H-purin-8-yl |
74 |
1,8a-dihydroindolizin-2-yl |
75 |
isoquinolin-6-yl |
|
76 |
quinolin-6-yl |
77 |
(cyclopenta-2,4-dien-1-yl)oxomethyl |
78 |
(2-methylcyclopenta-2,4-dien-1-yl)oxomethyl |
|
79 |
(2-fluorocyclopenta-2,4-dien-1-yl)oxomethyl |
80 |
(2-aminocyclopenta-2,4-dien-1-yl)oxomethyl |
81 |
(2-mercaptocyclopenta-2,4-dien-1-yl)oxomethyl |
|
82 |
(3-mercaptocyclopenta-2,4-dien-1-yl)oxomethyl |
83 |
(2,3-dimercaptocyclopenta-2,4-dien-1-yl)oxomethyl |
84 |
(2-chlorocyclopenta-2,4-dien-1-yl)oxomethyl |
|
85 |
(3-chlorocyclopenta-2,4-dien-1-yl)oxomethyl |
86 |
(2,3-dichlorocyclopenta-2,4-dien-1-yl)oxomethyl |
87 |
(3-bromocyclopenta-2,4-dien-1-yl)oxomethyl |
|
88 |
(2,3-dibromocyclopenta-2,4-dien-1-yl)oxomethyl |
89 |
(2-bromocyclopenta-2,4-dien-1-yl)oxomethyl |
90 |
(2-iodocyclopenta-2,4-dien-1-yl)oxomethyl |
|
91 |
(3-iodocyclopenta-2,4-dien-1-yl)oxomethyl |
92 |
(2,3-diiodocyclopenta-2,4-dien-1-yl)oxomethyl |
93 |
amino(cyclopenta-2,4-dien-1-yl)methyl |
|
94 |
amino(2-fluorocyclopenta-2,4-dien-1-yl)methyl |
95 |
amino(2,3-difluorocyclopenta-2,4-dien-1-yl)methyl |
96 |
amino(2-mercaptocyclopenta-2,4-dien-1-yl)methyl |
|
97 |
amino(2,3-dimercaptocyclopenta-2,4-dien-1-yl)methyl |
98 |
(2,3-dimercaptocyclopenta-2,4-dien-1-yl)(mercaptoamino)methyl |
99 |
(2-mercaptocyclopenta-2,4-dien-1-yl)(mercaptoamino)methyl |
|
100 |
(3-mercaptocyclopenta-2,4-dien-1-yl)(mercaptoamino)methyl |
101 |
(3-fluorocyclopenta-2,4-dien-1-yl)(mercaptoamino)methyl |
102 |
(2-fluorocyclopenta-2,4-dien-1-yl)(mercaptoamino)methyl |
|
103 |
(2,3-difluorocyclopenta-2,4-dien-1-yl)(mercaptoamino)methyl |
104 |
amino(2,3-dimercaptocyclopenta-2,4-dien-1-yl)methyl |
105 |
amino(2-mercaptocyclopenta-2,4-dien-1-yl)methyl |
|
106 |
(3-fluorocyclopenta-2,4-dien-1-yl)(fluoroamino)methyl |
107 |
(2,3-difluorocyclopenta-2,4-dien-1-yl)(fluoroamino)methyl |
108 |
(2,3-dichlorocyclopenta-2,4-dien-1-yl)(fluoroamino)methyl |
|
109 |
(2-chlorocyclopenta-2,4-dien-1-yl)(fluoroamino)methyl |
110 |
(3-chlorocyclopenta-2,4-dien-1-yl)(fluoroamino)methyl |
111 |
(3-bromocyclopenta-2,4-dien-1-yl)(fluoroamino)methyl |
|
112 |
(2,3-dibromocyclopenta-2,4-dien-1-yl)(fluoroamino)methyl |
113 |
(2-bromocyclopenta-2,4-dien-1-yl)(fluoroamino)methyl |
114 |
amino(2-aminooxomethylcyclopenta-2,4-dien-1-yl)methyl |
|
115 |
amino(3-aminooxomethylcyclopenta-2,4-dien-1-yl)methyl |
116 |
amino(2-aminooxomethyl-3-fluorocyclopenta-2,4-dien-1-yl)methyl |
117 |
amino(2-aminooxomethyl-3-chlorocyclopenta-2,4-dien-1-yl)methyl |
|
118 |
amino(2-aminooxomethyl-3-aminocyclopenta-2,4-dien-1-yl)methyl |
119 |
o-aminooxomethylphenyloxyoxomethyl |
120 |
m-aminooxomethylphenyloxyoxomethyl |
|
121 |
p-aminooxomethylphenyloxyoxomethyl |
122 |
o-mercaptophenyloxyoxomethyl |
123 |
m-mercaptophenyloxyoxomethyl |
|
124 |
p-mercaptophenyloxyoxomethyl |
125 |
m,o-dimercaptophenyloxyoxomethyl |
126 |
o-aminooxomethylphenyl imidmethyl |
|
127 |
phenyl imidmethyl |
128 |
m-aminooxomethylphenyl imidmethyl |
129 |
p-aminooxomethylphenyl imidmethyl |
|
130 |
o-mercaptophenyl imidmethyl |
131 |
m,o-dimercaptophenyl imidmethyl |
132 |
m-mercaptophenyl imidmethyl |
|
133 |
p-mercaptophenyl imidmethyl |
134 |
o-fluorophenyl imidmethyl |
135 |
m-fluorophenyl imidmethyl |
|
136 |
m-bromophenyl imidmethyl |
137 |
o-bromophenyl imidmethyl |
138 |
o-chlorophenyl imidmethyl |
|
139 |
m-chlorophenyl imidmethyl |
140 |
p-chlorophenyl imidmethyl |
141 |
2-chlorophenyl bromoimidmethyl |
|
142 |
4-chlorophenyl bromoimidmethyl |
143 |
4-bromophenyl bromoimidmethyl |
144 |
3-bromophenyl bromoimidmethyl |
|
145 |
2-bromophenyl bromoimidmethyl |
146 |
2-bromophenyl chloroimidmethyl |
147 |
3-bromophenyl chloroimidmethyl |
|
148 |
4-bromophenyl chloroimidmethyl |
149 |
4-chlorophenyl chloroimidmethyl |
150 |
3-chlorophenyl chloroimidmethyl |
|
151 |
2-chlorophenyl chloroimidmethyl |
152 |
2-methylphenyl imidmethyl |
153 |
3-methylphenyl imidmethyl |
|
154 |
4-methylphenyl imidmethyl |
155 |
4-(trifluoromethyl)phenyl imidmethyl |
156 |
3-(trifluoromethyl)phenyl imidmethyl |
|
157 |
2-(trifluoromethyl)phenyl imidmethyl |
158 |
2-(trifluoromethyl)phenyloxomethyl |
159 |
3-(trifluoromethyl)phenyloxomethyl |
|
160 |
4-(trifluoromethyl)phenyloxomethyl |
161 |
4-aminooxomethylphenyloxomethyl |
162 |
3-aminooxomethylphenyloxomethyl |
|
163 |
2-aminooxomethylphenyloxomethyl |
164 |
2-mercaptophenyloxomethyl |
165 |
3-mercaptophenyloxomethyl |
|
166 |
4-mercaptophenyloxomethyl |
167 |
3,4-dimercaptophenyloxomethyl |
168 |
2,3,4-trimercaptophenyloxomethyl |
|
169 |
2,4-dimercaptophenyloxomethyl |
170 |
2,5-dimercaptophenyloxomethyl |
171 |
2,4,5-trimercaptophenyloxomethyl |
|
172 |
2,3,5-trimercaptophenyloxomethyl |
173 |
2-methylphenyloxomethyl |
174 |
3-methylphenyloxomethyl |
|
175 |
4-methylphenyloxomethyl |
176 |
2-fluorophenyloxomethyl |
177 |
3-fluorophenyloxomethyl |
|
178 |
4-fluorophenyloxomethyl |
179 |
3,4-difluorophenyloxomethyl |
180 |
2,3,4-trifluorophenyloxomethyl |
|
181 |
2,4-difluorophenyloxomethyl |
182 |
2,3-difluorophenyloxomethyl |
183 |
2,3,4,5,6-pentafluorophenyloxomethyl |
|
184 |
1-(aminooxomethyl)-5-(amino(phosphino)methyl)cyclopenta-1,3-dien-2-yl |
185 |
aminooxomethyl |
186 |
4-chloro-1H-pyrazol-1-yl |
|
187 |
4,5-dichloro-1H-pyrazol-1-yl |
188 |
5-chloro-1H-pyrazol-1-yl |
189 |
3-chloro-1H-pyrazol-1-yl |
|
190 |
3,4,5-trichloro-1H-pyrazol-1-yl |
191 |
3-bromo-1H-pyrazol-1-yl |
192 |
4-bromo-1H-pyrazol-1-yl |
|
193 |
5-bromo-1H-pyrazol-1-yl |
194 |
4,5-dibromo-1H-pyrazol-1-yl |
195 |
3,4,5-tribromo-1H-pyrazol-1-yl |
|
196 |
4-mercapto-1H-pyrazol-1-yl |
197 |
4,5-dimercapto-1H-pyrazol-1-yl |
198 |
5-mercapto-1H-pyrazol-1-yl |
|
199 |
3,4,5-trimercapto-1H-pyrazol-1-yl |
200 |
1-iodo-1H-pyrazol-5-yl |
201 |
1-iodo-1H-pyrazol-4-yl |
|
202 |
1-iodo-1H-pyrazol-3-yl |
203 |
1,4-diiodo-1H-pyrazol-3-yl |
204 |
1,4,5-triiodo-1H-pyrazol-3-yl |
|
205 |
1,5-diiodo-1H-pyrazol-3-yl |
206 |
1,5-diiodo-1H-pyrazol-4-yl |
207 |
3,4,5-trifluoro-1H-pyrazol-1-yl |
|
208 |
5-fluoro-1H-pyrazol-1-yl |
209 |
4-fluoro-1H-pyrazol-1-yl |
210 |
3-fluoro-1H-pyrazol-1-yl |
|
211 |
3,4-difluoro-1H-pyrazol-1-yl |
212 |
3,5-difluoro-1H-pyrazol-1-yl |
213 |
3-amino-1H-pyrazol-1-yl |
|
214 |
4-amino-1H-pyrazol-1-yl |
215 |
5-amino-1H-pyrazol-1-yl |
216 |
4,5-diamino-1H-pyrazol-1-yl |
|
217 |
3,5-diamino-1H-pyrazol-1-yl |
218 |
3,4-diamino-1H-pyrazol-1-yl |
219 |
3,4,5-triamino-1H-pyrazol-1-yl |
|
220 |
5-methyl-1H-pyrazol-1-yl |
221 |
4-methyl-1H-pyrazol-1-yl |
222 |
3-methyl-1H-pyrazol-1-yl |
|
223 |
3,4-dimethyl-1H-pyrazol-1-yl |
224 |
3,5-dimethyl-1H-pyrazol-1-yl |
225 |
4,5-dimethyl-1H-pyrazol-1-yl |
|
226 |
3,4,5-trimethyl-1H-pyrazol-1-yl |
227 |
5-ethyl-1H-pyrazol-1-yl |
228 |
4-ethyl-1H-pyrazol-1-yl |
|
229 |
3-ethyl-1H-pyrazol-1-yl |
230 |
3,4-diethyl-1H-pyrazol-1-yl |
231 |
3,4,5-triethyl-1H-pyrazol-1-yl |
|
232 |
4,5-diethyl-1H-pyrazol-1-yl |
233 |
3,5-diethyl-1H-pyrazol-1-yl |
234 |
5-(mercaptomethyl)-1H-pyrazol-1-yl |
|
235 |
4-(mercaptomethyl)-1H-pyrazol-1-yl |
236 |
3-(mercaptomethyl)-1H-pyrazol-1-yl |
237 |
3,4-bis(mercaptomethyl)-1H-pyrazol-1-yl |
|
238 |
3,5-bis(mercaptomethyl)-1H-pyrazol-1-yl |
239 |
4,5-bis(mercaptomethyl)-1H-pyrazol-1-yl |
240 |
4-mercapto-5-(mercaptomethyl)-1H-pyrazol-1-yl |
|
241 |
5-(aminothio)-4-mercapto-1H-pyrazol-1-yl |
242 |
4,5-bis(aminothio)-1H-pyrazol-1-yl |
243 |
4,5-bis(aminothio)-3-mercapto-1H-pyrazol-1-yl |
|
244 |
5-ethyl-4-methyl-1H-pyrazol-1-yl |
245 |
5-ethyl-3,4-dimethyl-1H-pyrazol-1-yl |
246 |
phenyl |
|
247 |
pyridin-4-yl |
248 |
pyridazin-3-yl |
249 |
pyridazin-4-yl |
|
250 |
pyrimidin-4-yl |
251 |
1,3,5-triazin-2-yl |
252 |
pyrimidin-2-yl |
|
253 |
pyrazin-2-yl |
254 |
1,3,4-thiadiazol-2-yl |
255 |
1H-tetrazol-5-yl |
|
256 |
1H-pyrazol-1-yl |
257 |
7H-purin-2-yl |
258 |
7H-purin-6-yl |
|
259 |
7H-purin-7-yl |
260 |
1H-imidazol-1-yl |
261 |
1H-imidazol-5-yl |
|
262 |
1H-imidazol-4-yl |
263 |
1H-imidazol-2-yl |
264 |
9H-carbazol-1-yl |
|
265 |
9H-carbazol-2-yl |
266 |
9H-carbazol-3-yl |
267 |
9H-carbazol-4-yl |
|
268 |
1H-1,2,4-triazol-5-yl |
269 |
1H-1,2,3-triazol-1-yl |
270 |
1H-1,2,3-triazol-5-yl |
|
271 |
1H-1,2,3-triazol-4-yl |
272 |
anthracen-1-yl |
273 |
anthracen-2-yl |
|
274 |
acridin-1-yl |
275 |
acridin-2-yl |
276 |
acridin-3-yl |
|
277 |
isoxazol-3-yl |
278 |
isoxazol-4-yl |
279 |
isoxazol-5-yl |
|
280 |
1H-indol-7-yl |
281 |
1H-indol-6-yl |
282 |
1H-indol-5-yl |
|
283 |
1H-indol-4-yl |
284 |
1H-indol-3-yl |
285 |
1H-indol-2-yl |
|
286 |
1H-indol-1-yl |
287 |
thiophen-2-yl |
288 |
thiophen-3-yl |
|
289 |
thiazol-2-yl |
290 |
thiazol-5-yl |
291 |
pyrimidin-5-yl |
|
292 |
oxazol-2-yl |
293 |
oxazol-4-yl |
294 |
oxazol-5-yl |
|
295 |
furan-2-yl |
296 |
furan-3-yl |
297 |
thianthren-1-yl |
|
298 |
thianthren-2-yl |
299 |
indolizin-5-yl |
300 |
indolizin-6-yl |
|
301 |
indolizin-3-yl |
302 |
indolizin-2-yl |
303 |
indolizin-1-yl |
|
304 |
(oxomethyloxyl)dimethylethan-2-yl |
305 |
ureido |
306 |
isobutyl |
|
307 |
difluoromethyl |
308 |
4-hydrosulfonylphenyl |
309 |
tosyl |
|
310 |
mercapto |
311 |
(tetrahydro-2H-pyran-2-yl)oxyl |
312 |
6-hydroxytetrahydro-2H-pyran-2-yl |
|
313 |
6-hydroxytetrahydro-2H-pyran-3-yl |
314 |
2-hydroxytetrahydro-2H-pyran-4-yl |
315 |
hydrosulfinyl |
|
316 |
hydrosulfonyl |
317 |
hydrosulfonylamino |
318 |
sulfamoyl |
|
319 |
(dihydroxy)oxophosphyl hydroxyl |
320 |
hydroxyloxomethyl |
321 |
formyloxy |
|
322 |
(dihydroxy)oxophosphyl ethyl |
323 |
(methoxymethoxy)methyl |
324 |
(2-methoxyethoxy) methyl |
|
325 |
methyl-hydroxy-oxophosphyl methyl |
326 |
o-(oxomethyloxyl)methylphenyl |
327 |
m-(oxomethyloxyl)methylphenyl |
|
328 |
p-(oxomethyloxyl)methylphenyl |
329 |
benzyloxyoxomethyl |
330 |
benzyl |
|
331 |
o-tolyl |
332 |
m-tolyl |
333 |
p-tolyl |
|
334 |
oxophenylmethyl |
335 |
2-formylphenyl |
336 |
3-formylphenyl |
|
337 |
4-formylphenyl |
338 |
allyl |
339 |
prop-1-en-1-yl |
|
340 |
2-amino-2-oxoethyl |
341 |
acetamido |
342 |
cyclohexyl |
|
343 |
(2,4-dioxo-1,2,3,4-tetrahydropyrimidin-5-yl)methyl |
344 |
ethyl |
345 |
propyl |
|
346 |
butyl |
347 |
pentyl |
348 |
neopentyl |
|
349 |
piperidin-1-yl |
350 |
tetrahydropyridazin-1(2H)-yl |
351 |
piperazin-1-yl |
|
352 |
1,2,4-triazinan-1-yl |
353 |
4-benzylphenyl |
354 |
4-phenoxyphenyl |
|
355 |
5-benzylthiophen-2-yl |
356 |
5-(cyclohexylmethyl)thiophen-2-yl |
357 |
5-((4-methylpiperazin-1-yl)methyl)thiophen-2-yl |
|
358 |
5-((4-methylpiperazin-1-yl)methyl)furan-2-yl |
359 |
5-(4-(4-methylpiperazin-1-yl)phenyl)furan-2-yl |
360 |
5-(4-(4-methylpiperazin-1-yl)phenyl)thiophen-2-yl |
|
361 |
4-((4-methylpiperazin-1-yl)methyl)phenyl |
362 |
1,4-dioxin-2-yl |
363 |
4H-1,4-oxazin-3-yl |
|
364 |
3,4-dihydro-2H-1,2,4-oxadiazin-3-yl |
365 |
4-propylphenyl |
366 |
4-pentylphenyl |
|
367 |
4-butylphenyl |
368 |
4-isopropylphenyl |
369 |
hydro |
|
370 |
methyl |
371 |
isopropyl |
372 |
tert-butyl |
|
373 |
2-chlorobenzyl |
374 |
2-bromobenzyl |
375 |
2-nitrobenzyl |
|
376 |
3-chlorobenzyl |
377 |
3-bromobenzyl |
378 |
4-fluorobenzyl |
|
379 |
2-chloro-4-fluorobenzyl |
380 |
5-amino-2-chlorobenzyl |
381 |
4-fluoro-2-(trifluoromethyl)benzyl |
|
382 |
3-methoxybenzyl |
383 |
2-chloro-5-(2-phenylacetamido)benzyl |
384 |
2-chloro-5-(3-phenylureido)benzyl |
|
385 |
(6-methylpyridin-2-yl)methyl |
386 |
(6-methylpyridin-2-yl)thio |
387 |
((6-methylpyridin-2-yl)thio)methyl |
|
388 |
((6-methylpyridin-2-yl)oxy)methyl |
389 |
(5-methyl-2-(trifluoromethyl)furan-3-yl)methyl |
390 |
2-(3,5-dimethyl-1H-pyrazol-1-yl)ethyl |
|
391 |
(4-bromo-1-ethyl-1H-pyrazol-5-yl)methyl |
392 |
(3-methyl-1H-pyrazol-1-yl)methyl |
393 |
thiophen-2-ylmethyl |
|
394 |
(4-methylthiazol-2-yl)methyl |
395 |
(4,5-dimethylthiazol-2-yl)methyl |
396 |
fluoro |
|
397 |
hydroxyl |
398 |
chlorooxyl |
399 |
hydroxymethoxyl |
|
400 |
iodomethoxyl |
401 |
2-hydroxyl-2-oxoethoxyl |
402 |
2-amino-2-oxoethoxyl |
|
403 |
3-aminopropyl |
404 |
3-fluoropropyl |
405 |
4-oxobutyl |
|
406 |
trichloromethyl |
407 |
3,3,3-trifluoropropyl |
408 |
(E)-(hydroxydiazenyl)oxyl |
|
409 |
3,3-dihydroxyallyl |
410 |
(Z)-4-hydroxybut-2-en-1-yl |
411 |
(Z)-4-fluorobut-2-en-1-yl |
|
412 |
(Z)-4-cyanobut-2-en-1-yl |
413 |
5-hydroxyl-5-oxopentyl |
414 |
4-hydroxy-2-methylbutyl |
|
415 |
chloro |
416 |
bromooxyl |
417 |
chloromethoxyl |
|
418 |
mercaptomethoxyl |
419 |
2-oxoethoxyl |
420 |
nitromethoxyl |
|
421 |
3-hydroxypropyl |
422 |
3-iodopropyl |
423 |
3-cyanopropyl |
|
424 |
3-nitropropyl |
425 |
formyl |
426 |
2,2,2-trifluoroethyl |
|
427 |
2-fluoroethyl |
428 |
hydroxydiazenyl |
429 |
4-fluorobutyl |
|
430 |
3-hydroxyallyl |
431 |
4-aminobut-2-en-1-yl |
432 |
2,2-dihydroxyvinyl |
|
433 |
bromo |
434 |
iodo |
435 |
amino |
|
436 |
methoxyl |
437 |
iodooxyl |
438 |
mercaptooxyl |
|
439 |
cyano |
440 |
nitro |
441 |
trichloroethyl |
|
442 |
diazenyl |
443 |
nitroso |
444 |
trifluoromethyl |
|
445 |
vinyl |
||||
a All fragments were used for substitutions in the R1 and R2 positions.
3.5.2. In Silico Screening of Library of DAHAs
The focused library of 150,713 analogs was further screened for molecular structures matching the 3D-QSAR PH4 pharmacophore model Hypo1 of HDAC2 inhibition. 61 DAHAs mapped to at least 2 pharmacophoric features and 49 of which mapped to at least 3 features of the pharmacophore. These 110 best fitting analogs (PH4 hits) then underwent complexation QSAR model screening. The computed GFE of HDAC2-DAHAx complex formation, their components, and predicted half-maximal inhibitory concentrations IC50pre calculated from the correlation Equation (B) (Table 3) are listed in Table 6.
Table 6. GFE and their components for the top-scoring 110 virtual DAHA analogs. The analog numbering concatenates the index of each substituent R1 to R2 with the substituent numbers taken from Table 5.
|
Designed Analogs |
∆∆HMM a | ∆∆Gsol b |
∆∆TSvib c | ∆∆Gcom d | IC50pre e |
|
|
[kcal.mol-1] |
[kcal.mol-1] |
[kcal.mol-1] |
[kcal.mol-1] |
[nM] |
||
|
N° |
DAHA1 |
0.0 |
0.0 |
0.0 |
0.0 |
260.0 |
|
1 |
1-109 |
-3.5 |
0.8 |
-4.9 |
2.2 |
2733.4 |
|
2 |
1-114 |
-6.4 |
2.9 |
-2.9 |
-0.6 |
203.4 |
|
3 |
1-410 |
-4.4 |
0.5 |
0.0 |
-3.9 |
9.6 |
|
4 |
2-210 |
-2.9 |
1.5 |
-4.0 |
2.6 |
4035.0 |
|
5 |
2-438 |
-2.4 |
-0.3 |
-5.2 |
2.6 |
3874.0 |
|
6 |
3 -97 |
-5.2 |
2.0 |
-1.0 |
-2.2 |
47.2 |
|
7 |
3-191 |
-3.2 |
2.0 |
-5.4 |
4.2 |
17365.0 |
|
8 |
3-403 |
-4.6 |
-0.7 |
1.0 |
-6.4 |
1.0 |
|
9 |
3-395 |
-6.0 |
0.9 |
-2.4 |
-2.8 |
27.4 |
|
10 |
3-430 |
-5.5 |
-0.5 |
-0.9 |
-5.1 |
3.2 |
|
11 |
4-197 |
-5.9 |
0.0 |
-3.8 |
-2.1 |
51.7 |
|
12 |
4-215 |
-3.8 |
2.4 |
-3.2 |
1.8 |
1972.0 |
|
13 |
4-241 |
-1.8 |
1.2 |
-5.0 |
4.3 |
19738.0 |
|
14 |
5-42 |
-14.4 |
0.8 |
-9.5 |
-4.1 |
8.3 |
|
15 |
5-262 |
-7.7 |
2.2 |
-6.3 |
0.9 |
801.2 |
|
16 |
166-239 |
-4.8 |
1.9 |
-3.4 |
0.6 |
617.1 |
|
17 |
5-413 |
-1.9 |
-0.4 |
0.5 |
-2.8 |
27.8 |
|
18 |
166-418 |
-5.6 |
1.0 |
-6.5 |
2.0 |
2212.0 |
|
19 |
177-99 |
-8.2 |
1.9 |
-3.2 |
-3.2 |
19.9 |
|
20 |
175-277 |
-7.3 |
1.0 |
-3.2 |
-3.0 |
21.9 |
|
21 |
174-101 |
-0.7 |
-2.0 |
2.2 |
-4.8 |
4.2 |
|
22 |
174-234 |
-0.1 |
-1.7 |
-1.5 |
-0.2 |
294.0 |
|
23 |
173-338 |
-3.1 |
-2.5 |
-2.4 |
-3.2 |
18.4 |
|
24 |
5-409 |
-5.5 |
0.7 |
-2.7 |
-2.1 |
52.6 |
|
25 |
43-409 |
-1.8 |
2.0 |
2.5 |
-2.2 |
45.9 |
|
26 |
43-410 |
-2.0 |
1.2 |
3.6 |
-4.4 |
6.1 |
|
27 |
43-294 |
-6.2 |
1.0 |
-1.4 |
-3.8 |
11.1 |
|
28 |
6-118 |
-5.6 |
3.9 |
-2.7 |
1.1 |
953.0 |
|
29 |
6-403 |
-5.0 |
0.5 |
0.3 |
-4.9 |
4.0 |
|
30 |
6-357 |
-9.4 |
2.0 |
-4.0 |
-3.3 |
17.4 |
|
31 |
7-318 |
-8.7 |
3.4 |
-9.1 |
3.8 |
12109.0 |
|
32 |
7-401 |
-8.8 |
1.2 |
-5.7 |
-1.9 |
63.2 |
|
33 |
8-95 |
-8.2 |
-0.9 |
-2.4 |
-6.7 |
0.8 |
|
34 |
44-259 |
-7.3 |
2.1 |
-2.7 |
-2.5 |
36.1 |
|
35 |
46-239 |
-6.2 |
-0.6 |
-2.2 |
-4.6 |
5.4 |
|
36 |
49-345 |
-3.6 |
-1.0 |
2.7 |
-7.2 |
0.5 |
|
37 |
59-410 |
-4.1 |
0.8 |
0.0 |
-3.4 |
16.6 |
|
38 |
8-222 |
-4.2 |
2.1 |
-3.9 |
1.8 |
1932.0 |
|
39 |
8-234 |
-6.3 |
0.1 |
-4.4 |
-1.9 |
64.7 |
|
40 |
8-421 |
-3.3 |
1.4 |
-0.9 |
-1.1 |
138.1 |
|
41 |
8-410 |
-8.2 |
2.5 |
0.1 |
-5.8 |
1.8 |
|
42 |
9-235 |
-0.5 |
2.4 |
-2.2 |
4.1 |
15955.0 |
|
43 |
9-113 |
-10.2 |
4.0 |
-3.3 |
-2.9 |
26.3 |
|
44 |
9-358 |
-3.5 |
0.5 |
-4.0 |
1.0 |
883.0 |
|
45 |
68-93 |
-1.1 |
-0.4 |
-0.7 |
-0.7 |
183.8 |
|
46 |
71-364 |
-6.4 |
1.6 |
3.0 |
-7.8 |
0.3 |
|
47 |
10-238 |
-3.0 |
-0.4 |
-2.9 |
-0.5 |
235.1 |
|
48 |
10-248 |
-5.9 |
0.7 |
-5.0 |
-0.2 |
290.8 |
|
49 |
10-350 |
-3.1 |
-1.2 |
1.5 |
-5.8 |
1.8 |
|
50 |
11-239 |
-5.9 |
-1.4 |
-5.1 |
-2.2 |
48.6 |
|
51 |
11-242 |
-6.1 |
-3.6 |
-7.8 |
-1.9 |
61.8 |
|
52 |
11-254 |
-6,6 |
0.8 |
-9.0 |
3.1 |
6206.0 |
|
53 |
76-397 |
-0.5 |
-0.2 |
-3.2 |
2.5 |
3767.0 |
|
54 |
75-296 |
-3.3 |
0.5 |
-5.4 |
2.6 |
4012.0 |
|
55 |
79-314 |
-1.3 |
-1.6 |
-1.4 |
-1.5 |
92.6 |
|
56 |
79-33 |
-9.4 |
-1.4 |
-6.0 |
-4.9 |
3.9 |
|
57 |
17-432 |
-5.0 |
3.4 |
-2.1 |
0.5 |
585.2 |
|
58 |
18-351 |
-4.4 |
-1.6 |
-0.4 |
-5.6 |
2.0 |
|
59 |
19-295 |
-1.6 |
2.5 |
-2.9 |
3.8 |
12097.0 |
|
60 |
19-431 |
-3.3 |
1.6 |
-0.6 |
-1.1 |
134.0 |
|
61 |
19-350 |
-5.7 |
1.7 |
-0.9 |
-3.1 |
21.3 |
|
62 |
22-398 |
-15.5 |
6.5 |
-6.9 |
-2.2 |
49.4 |
|
63 |
23-41 |
-10.9 |
8.2 |
-4.8 |
2.0 |
2350.0 |
|
64 |
13-242 |
-10.8 |
2.8 |
-6.9 |
-1.1 |
129.7 |
|
65 |
13-394 |
-14.7 |
7.2 |
-3.5 |
-4.0 |
9.4 |
|
66 |
31-245 |
-6.1 |
1.2 |
-1.3 |
-3.6 |
13.4 |
|
67 |
31-399 |
-5.2 |
2.3 |
-6.3 |
3.4 |
8126.0 |
|
68 |
32-263 |
-12.0 |
4.4 |
-6.4 |
-1.2 |
119.5 |
|
69 |
32-435 |
-9.8 |
2.3 |
-6.6 |
-0.9 |
154.9 |
|
70 |
33-227 |
-3.3 |
0.3 |
0.0 |
-3.0 |
23.5 |
|
71 |
28-433 |
-10.4 |
1.2 |
-10.1 |
1.0 |
895.4 |
|
72 |
15-311 |
-2.7 |
-0.3 |
3.3 |
-6.3 |
1.1 |
|
73 |
33-394 |
-10.2 |
1.8 |
-4.5 |
-3.9 |
9.7 |
|
74 |
34-394 |
-10.1 |
2.6 |
-4.9 |
-2.6 |
33.5 |
|
75 |
36-217 |
-10.2 |
6.2 |
-0.6 |
-3.3 |
17.3 |
|
76 |
37-416 |
-8.6 |
4.0 |
-6.5 |
1.8 |
1989.0 |
|
77 |
39-399 |
-8.8 |
5.5 |
-1.6 |
-1.7 |
76.5 |
|
78 |
40-418 |
-8.3 |
3.7 |
-3.5 |
-1.0 |
140.4 |
|
79 |
95-221 |
-3.9 |
-2.8 |
-0.4 |
-6.3 |
1.1 |
|
80 |
97-333 |
0.4 |
-0.4 |
-0.1 |
0.0 |
352.0 |
|
81 |
103-359 |
-7.0 |
-0.4 |
-0.2 |
-7.2 |
0.5 |
|
82 |
102-421 |
1.2 |
-0.7 |
0.3 |
0.2 |
428.8 |
|
83 |
118-307 |
-4.9 |
-0.6 |
-2.2 |
-3.4 |
16.5 |
|
84 |
16-437 |
-7.1 |
-3.1 |
-7.8 |
-2.5 |
37.7 |
|
85 |
182-407 |
-5.9 |
-3.5 |
-8.0 |
-1.4 |
98.7 |
|
86 |
178-99 |
-3.3 |
-1.5 |
-4.6 |
-0.2 |
289.7 |
|
87 |
180-103 |
-5.8 |
0.6 |
-6.8 |
1.6 |
1618.0 |
|
88 |
196-126 |
1.4 |
0.0 |
-2.1 |
3.5 |
9282.0 |
|
89 |
200-115 |
-2.5 |
1.1 |
-4.7 |
3.3 |
7467.0 |
|
90 |
441-388 |
-1.4 |
-2.3 |
-4.2 |
0.5 |
596.6 |
|
91 |
191-153 |
-8.0 |
1.1 |
-5.2 |
-1.7 |
79.2 |
|
92 |
428-390 |
-1.0 |
-1.5 |
-1.1 |
-1.4 |
105.0 |
|
93 |
426-284 |
2.6 |
-2.7 |
0.4 |
-0.5 |
239.3 |
|
94 |
430-72 |
-1.0 |
-3.0 |
-4.0 |
-0.1 |
345.6 |
|
95 |
437-152 |
-0.9 |
-2.9 |
-5.6 |
1.8 |
1980.0 |
|
96 |
421-299 |
2.1 |
1.1 |
1.8 |
1.5 |
1408.0 |
|
97 |
382-318 |
1.2 |
1.8 |
4.8 |
-1.8 |
67.7 |
|
98 |
368-431 |
1.2 |
-2.0 |
6.7 |
-7.5 |
0.4 |
|
99 |
368-371 |
0.0 |
-1.7 |
4.8 |
-6.4 |
1.0 |
|
100 |
295-417 |
0.6 |
-2.2 |
0.2 |
-1.7 |
73.0 |
|
101 |
204-296 |
-3.6 |
-6.0 |
-8.5 |
-1.1 |
138.3 |
|
102 |
301-239 |
0.8 |
-0.4 |
1.5 |
-1.1 |
135.2 |
|
103 |
301-212 |
-2.7 |
-1.9 |
-3.8 |
-0.8 |
173.0 |
|
104 |
313-75 |
-1.2 |
-0.9 |
0.7 |
-2.8 |
27.8 |
|
105 |
323-129 |
-1.4 |
1.7 |
0.8 |
-0.5 |
232.0 |
|
106 |
333-238 |
1.0 |
-1.5 |
-2.2 |
1.7 |
1790.0 |
|
107 |
337-288 |
-4.4 |
-0.4 |
-3.5 |
-1.3 |
111.4 |
|
108 |
344-381 |
-0.5 |
-7.0 |
-3.7 |
-3.8 |
11.4 |
|
109 |
352-284 |
-4.6 |
3.1 |
2.7 |
-4.1 |
8.1 |
|
110 |
364-377 |
-2.1 |
0.6 |
-0.3 |
-1.2 |
120.0 |
a ∆∆HMM is the relative enthalpic contribution to the GFE change of the HDAC2-DAHA complex formation ∆∆Gcom (for details see footnote of Table 2); b ∆∆Gsol is the relative solvation GFE contribution to ∆∆Gcom ; c ∆∆TSvib is the relative (vibrational) entropic contribution to ∆∆Gcom ; d ∆∆Gcom is the relative Gibbs free energy change related to the enzyme–inhibitor HDAC2-DAHA complex formation ∆∆Gcom ≅ ∆∆HMM + ∆∆Gsol - ∆∆TSvib; e IC50pre is the predicted inhibition potency towards HDAC2 calculated from ∆∆Gcom using correlation Equation (B), Table 3; IC50exp is given for the reference inhibitor DAHA1 instead of the IC50pre.
3.6. Analysis of novel DAHA analogs substituents
To identify which substituents on R-positions of DAHA scaffold (Table 5) lead to new inhibitor candidates with the highest predicted potencies towards the HDAC2, histograms of the absolute frequency of occurrence of R1- and R2- groups among the 110 best fit PH4 hits were prepared (Figure 6). From these histograms, it comes out that R1-groups numbered 3(5), 8(5), 5(4), 1(3), 4(3), 6(3), 9(3), 10(3), 11(3), 19(3), and 43(3) are almost equally represented with the highest occurrence in DAHAS subset. The R2-groups contain preferentially 239(4), 410(4), and 394(3).
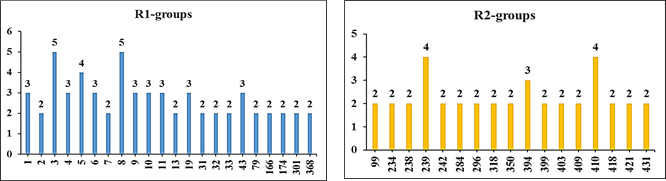
Figure 6. Histograms of frequency of occurrence of individual R-groups in the 110 best-selected analogs mapping to features of the PH4 pharmacophore hypothesis Hypo1 (for the structures of the fragments see Table 5).
3.7. ADME Profile of Novel DAHA Analogs
The properties related to ADME such as octanol-water partitioning coefficient, aqueous solubility, blood-brain partition coefficient, Caco-2 cell permeability, serum protein binding, number of likely metabolic reactions, and another eighteen descriptors related to absorption, distribution, metabolism, and excretion (ADME) were calculated by the QikProp program [31] for the new best DAHA analogs (Table 7). This program is based on the method of Jorgensen [38, 39]. Experimental data from more than 710 compounds including about 500 drugs and related heterocycles were used to produce regression equations correlating experimental and computed descriptors resulting in an accurate prediction of pharmacokinetic properties of molecules. Drug likeness (#stars) - the number of property descriptors that fall outside the range of optimal values determined for 95% of known drugs out of 24 selected descriptors computed by the QikProp, was used as an additional ADME-related compound selection criterion. The values for the best active designed DAHAs are compared with those computed for drugs used for the treatment of cancer or currently undergoing clinical trials, Table 7.
Table 7. Predicted ADME-related properties of the best designed DAHA analogs and known anticancer agents either in clinical use or currently undergoing clinical testing computed by QikProp [31].
|
a designed DAHA analogs and known anticancer agents, Table 6; b drug likeness, number of property descriptors (24 out of the full list of 49 descriptors of QikProp, ver. 3.7, release 14) that fall outside of the range of values for 95% of known drugs; c molar mass in [g.mol-1] (range for 95% of drugs: 130–725 g.mol−1) [31] ; d total solvent-accessible molecular surface, in [Å2] (probe radius 1.4 Å) (range for 95% of drugs: 300–1000 Å2); e hydrophobic portion of the solvent-accessible molecular surface, in [Å2] (probe radius 1.4 Å) (range for 95% of drugs: 0–750 Å2); f total volume of molecule enclosed by solvent-accessible molecular surface, in [Å3] (probe radius 1.4 Å) (range for 95% of drugs: 500–2000 Å3); g number of non-trivial (not CX3), non-hindered (not alkene, amide, small ring) rotatable bonds (range for 95% of drugs: 0–15); h estimated number of hydrogen bonds that would be donated by the solute to water molecules in an aqueous solution. Values are averages taken over several configurations, so they can assume non-integer values (range for 95% of drugs: 0.0–6.0); i estimated the number of hydrogen bonds that would be accepted by the solute from water molecules in an aqueous solution. Values are averages taken over a number of configurations, so they can assume non-integer values (range for 95% of drugs: 2.0–20.0); j logarithm of partitioning coefficient between n-octanol and water phases (range for 95% of drugs: −2 to 6.5); k logarithm of predicted aqueous solubility, logS. S in [mol·dm–3] is the concentration of the solute in a saturated solution that is in equilibrium with the crystalline solid (range for 95% of drugs: −6.0 to 0.5); l logarithm of predicted binding constant to human serum albumin (range for 95% of drugs: −1.5 to 1.5); m logarithm of predicted brain/blood partition coefficient (range for 95% of drugs: −3.0 to 1.2); n predicted apparent Caco-2 cell membrane permeability in Boehringer-Ingelheim scale in [nm s-1] (range for 95% of drugs: < 25 poor, > 500 nm s−1 great); o number of likely metabolic reactions (range for 95% of drugs: 1–8); p predicted inhibition constants IC50pre was predicted from computed ∆∆Gcom using the regression Equation (B) shown in Table 3; q human oral absorption (1 = low, 2 = medium, 3 = high); r percentage of human oral absorption in gastrointestinal tract (<25% = poor, >80% = high); * star in any column indicates that the property descriptor value of the compound falls outside the range of values for 95% of known drugs.
4.1. Binding mode of new inhibitors from in Silico screening
An analysis of structural requirements for human HDAC2 inhibition at the high affinity of the amine-based hydroxamic acid derivatives with the active site revealed that the substituents at the R2 position in the training set are large. Therefore, new DAHA analogs that match the HDAC2 inhibition pharmacophore and fill better the site II (S2 sub-pocket) may form potent HDAC2 inhibitors (Table 6). The top-scoring virtual hits are DAHA analogs: 49-345 (IC50pre =0.5 nM), 368-371(IC50pre =1.0 nM), 95-221(IC50pre =1.1 nM), 10-350(IC50pre =1.8 nM) and 3-430(IC50pre =3.2 nM). The best analog designed 49-345 (IC50pre =0.5 nM) displays predicted potency approximately 520 times better than the best training set compound DAHA1 (IC50exp = 260 nM). The approach taken in this work helped to identify interesting R1-groups such as 4 - (3,4,5-trimethyl-1H-pyrazol-1-yl) phenyl (49), 4 - isopropyl phenyl (368), amino (2,3-difluorocyclopenta-2,4-dien-1-yl) methyl (95), 4 - (5-bromo-1H-pyrazol-1-yl) (10), and 4 - (3-fluoro-1H-pyrazol-1-yl) (3) for the filling of the S1 sub-pocket with a bulkier group compared to the training set inhibitors.
The same approach made it possible to identify interesting R2-groups, which are least bulky but most specific to the sub-pocket of site II such as propyl (345), isoprpyl (371), 4 -methyl-1H-pyrazol-1-yl (221), tetrahydropyridazin-1(2H)-yl (350), and 3-hydroxyallyl (430) Table 5.
Analysis of the HDAC2-DAHAx complexes of the most potent inhibitors shows that several interactions play a key role in the significant improvement of the predicted inhibitory potencies of the novel amine-based hydroxamic acid derivatives.
According to Pavel et al. [12], one of the main characteristics of the binding site in HDAC class I and II isoforms is an aspartic amino acid Asp104 (in HDAC2, a different number in other HDACs) located at the gorge region of the binding site. The work of Sanjay K. Choubey & Jeyaraman Jeyakanthan [40] showed that Glu103 and Asp104 are likely to form hydrogen bonds with HDAC2 in inhibitors which is consistent with the data of our study. Indeed, the best compound (49-345) demonstrated a better binding affinity with the residues of the active site of HDAC2 (figure 7. c,d). The ZBG of 49-345 retains hydrogen bonds with His145, Asp181, His183, and Tyr308 (Figure 7. c,d). The linker well stabilized in the hydrophobic channel formed by Gly154, Phe155, Phe210, and Leu276. The R1 substituent [4-(3,4,5-trimethyl-1H-pyrazol-1-yl) phenyl] can pinch site I residues such as Asp104 via a Pi-Anion and a hydrogen bond, thus resembling the known HDAC2 squaramide inhibitor [41]; Arg275 via a Pi-Donor Hydrogen Bond; His33 and Pro34 via Alkyl interactions; Gly32 and Glu103 via van der Waals interactions (Figure 7. c,d). The R2 substituent (propyl) clamps the hydrophobic residue Leu276 from site II via an Alkyl interaction (Figure 7. a,c,d). This Leu276 residue located in the hydrophobic channel (sub-pocket S2) leaves more space to accommodate the hydrophobic groups propyl (345) and isopropyl (371) which is consistent with the increased potency of top analogs 49-345 and 368- 371 to inhibit HDAC2 activity.
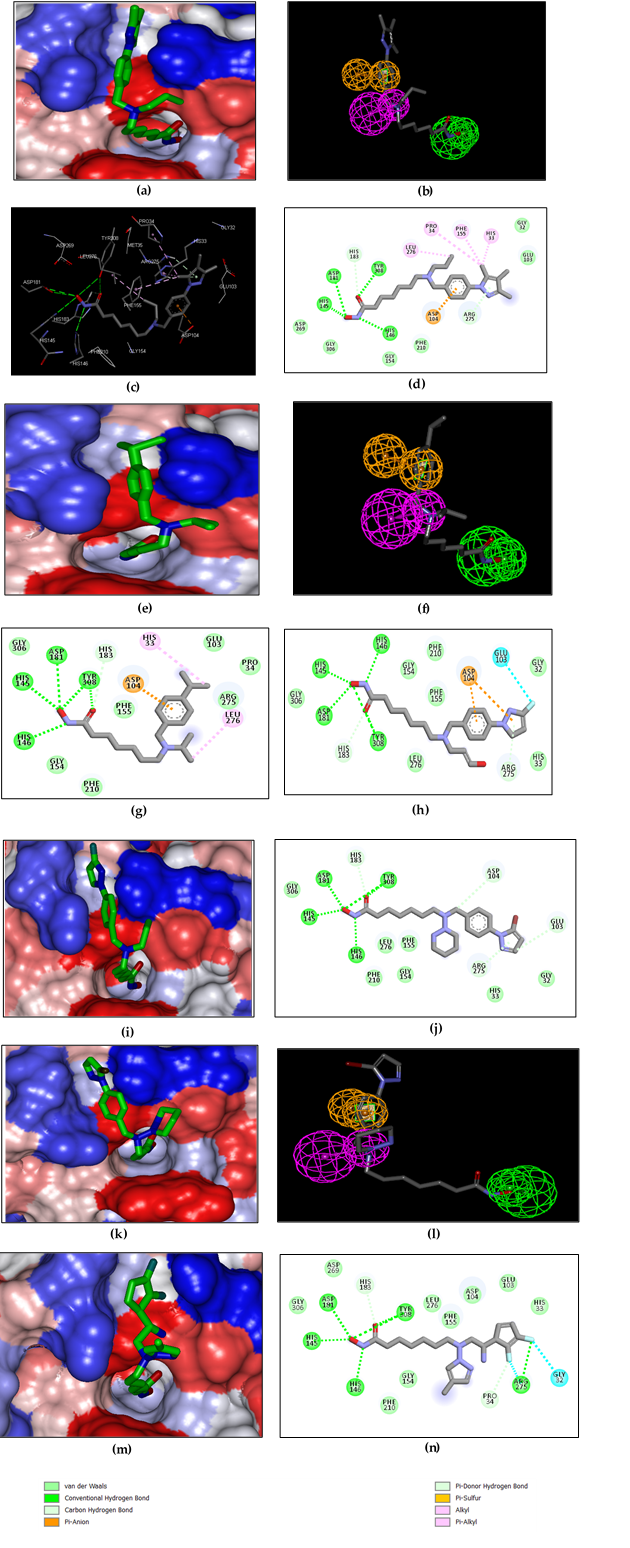
Figure 7. (a) Connolly surface of the active site of HDAC2 with bound most active designed DAHA analog 49-345 (IC50pre =0.5 nM). The binding site surface is coloured according to residue hydrophobicity: red - hydrophobic, blue - hydrophilic and white – intermediate; (b) mapping of the DAHA 49-345 to HDAC2 inhibition pharmacophore; (c) close up of virtual hit DAHA 49-345 at the active site of HDAC2; (d) 2D schematic interaction diagram of the DAHA 49-345 at the active site of HDAC2; (e) Connolly surface of the active site of HDAC2 with bound DAHA analog 368-371(IC50pre =1.0 nM);(f) mapping of the DAHA 368-371 to HDAC2 inhibition pharmacophore; (g) 2D schematic interaction diagram of the analog DAHA 368-371 (IC50pre =1.0 nM) at the active site of HDAC2; (h) 2D schematic interaction diagram of the analog DAHA 3-430(IC50pre =3.2 nM) at the active site of HDAC2; (i) Connolly surface of the active site of HDAC2 with bound DAHA analog 3-430(IC50pre =3.2 nM); (j) 2D schematic interaction diagram of the analog DAHA 10-350(IC50pre =1.8 nM) at the active site of HDAC2; (k) Connolly surface of the active site of HDAC2 with bound DAHA analog 10-350(IC50pre =1.8 nM);(l) mapping of the DAHA 10-350 to HDAC2 inhibition pharmacophore; (m) Connolly surface of the active site of HDAC2 with bound DAHA analog 95-221(IC50pre =1.1 nM);(n) 2D schematic interaction diagram of the analog DAHA 95-221 (IC50pre =1.1 nM)at the active site of HDAC2.
4.2. Interaction Energy of new inhibitors from in Silico screening
According to our analysis of the HDAC2-DAHAx complexes of the most potent inhibitors, several interactions play a key role in significantly improving the predicted inhibitory powers of new amine-based hydroxamic acid derivatives. Based on the distribution of intermolecular interaction energy to residue contributions (Figure 8), residues Glu103, Asp104, Arg275, Leu276; in addition to the catalytic residues His145, His146, Phe155, Asp181, Phe210, His183, Tyr308 play a key role in the inhibition of HDAC2.
Indeed, FIG. 8 shows the increase in the affinity, through interaction energy between the charged residues Glu103, Asp104 of the sub-pocket S1 and the fragments (49): 4 - (3,4,5-trimethyl-1H-pyrazol-1-yl) phenyl; (95): amino (2,3-difluorocyclopenta-2,4-dien-1-yl) methyl; (10): 4 - (5-bromo-1H-pyrazol-1-yl) phenyl; (3): 4- (3-fluoro-1H-pyrazol-1-yl) phenyl and (71): 4 - (piperazin-1-yl) phenyl of five of the best-designed analogs 49-345(IC50pre =0.5 nM);95-221(IC50pre =1.1 nM);10-350(IC50pre =1.8 nM);3-430(IC50pre =3.2 nM) and 71-364(IC50pre =0.3 nM)compared to the most active training set inhibitor DAHA1 (IC50exp =260 nM). The residue charged Arg275 of the sub-pocket S1 also shows a strong energy connection with the fragments (3): 4-(3-fluoro-1H-pyrazol-1-yl)phenyl and (71): 4-(piperazin-1 -yl)phenyl of analogs 3-430 and 71-364 respectively compared to the most active training set inhibitor DAHA1.
The interaction energy contribution of Leu276 residue of the sub-pocket S2 is significant for the best six designed novel DAHA analogs compared to the DAHA1 inhibitor of the training set (see FIG. 8).
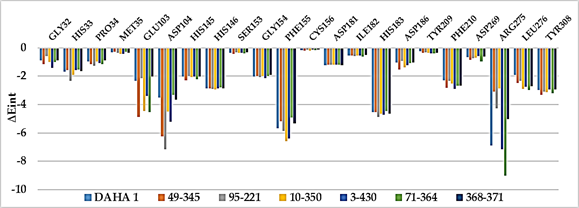
Figure 8. Molecular mechanics intermolecular interaction energy Eint breakdown to residue contributions, in [kcal.mol-1] shown for the best six designed novel DAHA analogs (the color coding refers to ligands and is given in the legend).
Design of new potent DAHA analogs inhibiting human HDAC2 with favorable pharmacokinetic profiles needed to extend the portfolio of currently available anticancer drugs. Structural information from the crystal structure of the HDAC2-SAHA complex guided us during the development of a reliable QSAR model for the non-covalent inhibition of HDAC2 by amine-based hydroxamic acid derivatives (DAHA), which correlated the computed Gibbs free energies of complex formation with the observed HDAC2 inhibitory potencies [12]. In addition to this QSAR model, we have elaborated a 3D QSAR pharmacophore model for DAHA inhibitors. Analysis of interactions between HDAC2 and DAHA in the active site of the enzyme was helpful in our effort to design a virtual combinatorial library (VCL) of new DAHA analogs with two substitutions (R1 and R2-groups). The initial virtual library was screened by matching PH4 pharmacophore analogs and allowed the selection of a focused library subset. The best virtual compounds were subjected to the prediction of inhibitory potencies from computed GFE through the QSAR model derived from the training set of known DAHAs. The best-designed analogs display predicted low nanomolar inhibitory concentrations 49-345(IC50pre =0.5 nM); 368-371(IC50pre =1.0 nM);95-221(IC50pre =1.1 nM); 10-350(IC50pre =1.8 nM); 3-430(IC50pre =3.2 nM); 46-239(IC50pre =5.4 nM); 43-410(IC50pre =6.1 nM) (Table 6). The predicted inhibitory potencies of the best-designed analogs are up to 520-fold higher than that of the most active training set inhibitor DAHA1. They are recommended for synthesis and biological evaluation to specialized laboratories to develop new anticancer drugs with a promising pharmacokinetic profile.
The authors thank Eugene Megnassan for his helpful advice.
Abbreviations
|
IC50 |
Half-maximal inhibitory concentration |
||
|
IE |
Interaction energy |
||
|
LOO |
Leave-one-out cross-validation |
||
|
MM |
Molecular mechanics |
||
|
PDB |
Protein Data Bank |
||
|
PH4 |
Pharmacophore |
||
|
PTCL |
Peripheral T-cell lymphoma |
||
|
PTM |
Post-translational modifications |
||
|
QSAR |
Quantitative structure-activity relationships |
||
|
RMSD |
Root-mean square deviation |
||
|
SBG |
Surface binding group |
||
|
TS |
Training set |
||
|
VLC |
Virtual combinatorial library |
||
|
VS |
Validation set |
||
|
ZBG |
Zinc binding group |
||
Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; et al. Global Cancer Observatory: Cancer Today. Lyon: International Agency for Research on Cancer; 2020. Available online: (accessed on 12 August 2021).
View ArticleNicholson, J. M.; Wood, C.M.; Reynolds, C.D.; Brown, A.; Lambert, S.J.; Chantalat, L. et Baldwin, J.P. Histone structures: targets for modifications by molecular assemblies. Ann N Y Acad Sci. 2004, 1030, 644-55. PMid:15659848
View Article PubMed/NCBIStrahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nat. 2000, 403, 41-5. PMid:10638745
View Article PubMed/NCBIPark, S.Y.; Kim, J.S. A short guide to histone deacetylases including recent progress on class II enzymes. Exp. Mol. Med. 2020, 52, 204-212. PMid:32071378
View Article PubMed/NCBISanaei, M.; Kavoosi, F. Histone deacetylases, and histone deacetylase inhibitors: Molecular mechanisms of action in various cancers. Adv. Biomed. Res. 2019, 8, 63. PMid:31737580
View Article PubMed/NCBIBondarev, A.D.; Attwood, M.M.; Jonsson, J.; Chubarev, V.N.; Tarasov, V.V.; Schiöth, H.B. Recent developments of HDAC inhibitors: Emerging indications and novel molecules. Br. J. Clin. Pharmacol. 2021, 1-21. PMid:33971031
View Article PubMed/NCBIKim, T.Y.; Bang, Y.J.; Robertson, K.D. Histone Deacetylase Inhibitors for Cancer Therapy. Epigenetics. 2006, 1, 14-23. PMid:17998811
View Article PubMed/NCBIRiester, D.; Hildmann, C.; Schwienhorst, A. Histone deacetylase inhibitors-turning epigenetic mechanisms of gene regulation into tools of therapeutic intervention in malignant and other diseases. Appl. Microbiol. Biotechnol. 2007, 75, 499-514. PMid:17377788
View Article PubMed/NCBIBressi, J.C.; Jennings, A.J.; Skene, R.; Wu, Y.; Melkus, R.; De Jong, R.; O'Connell, S.; Grimshaw, C.E.; Navre, M.; Gangloff, A.R. Exploration of the HDAC2 foot pocket: Synthesis and SAR of substituted N-(2 aminophenyl) benzamides. Bioorg. Med. Chem. Lett. 2010, 20, 3142-3145. PMid:20392638
View Article PubMed/NCBINoor, Z.; Afzal, N.; Rashid, S. Exploration of Novel Inhibitors for Class I Histone Deacetylase Isoforms by QSAR Modeling and Molecular Dynamics Simulation Assays. PLoS ONE. 2015, 10, e0139588. PMid:26431201
View Article PubMed/NCBILauffer, B.E.L.; Mintzer, R.; Fong, R.; Mukund, S.; Tam, C.; Zilberleyb, I.; … Steiner, P. Histone Deacetylase (HDAC) Inhibitor Kinetic Rate Constants Correlate with Cellular Histone Acetylation but Not Transcription and Cell Viability. J. Biol. Chem. 2013, 288, 26926-26943. PMid:23897821
View Article PubMed/NCBIPavel, A.P.; Hazem, A.; Raghupathi, N.; Antonett, M.; Irida, K.; Yue-ting, W.; Aditya, S.V.; Taha, Y.T.; Gregory, R.J.T.; Jonna, F. Design, Synthesis, Molecular Modeling, and Biological Evaluation of Novel Amine-based Histone Deacetylase Inhibitors. Chem. Med. Chem. 2017, 12, 2030-2043. PMid:29080240
View Article PubMed/NCBIInsight-II and Discover Molecular Modeling and Simulation Package, version 2005; Accelrys: San Diego, CA, USA, 2005.
Kouassi, A.F.; Kone, M.; Keita, M.; Esmel, A.; Megnassan, E.; N'Guessan, Y.T.; Frecer, V.; Miertus, S. Computer-aided design of orally bioavailable pyrrolidine carboxamide inhibitors of Enoyl-Acyl Carrier Protein Reductase of Mycobacterium tuberculosis with favorable pharmacokinetic profiles. Int. J. Mol. Sci. 2015, 16, 29744-29771. PMid:26703572
View Article PubMed/NCBIAllangba, K.N.P.G.; Keita, M.; KreN'Guessan, R.; Megnassan, E.; Frecer,V.; Miertus, S. Virtual design of novel Plasmodium falciparum cysteine protease falcipain-2 hybrid lactone-chalcone and isatin-chalcone inhibitors probing the S2 active site pocket. J. Enz. Inhib. Med. Chem. 2018, 34, 547-561. PMid:30696325
View Article PubMed/NCBIOwono Owono, L.C.; Ntie-Kang, F.; Keita, M.; Megnassan, E.; Frecer, V.; Miertus, S. Virtually designed triclosan-based inhibitors of enoyl-acyl carrier protein reductase of Mycobacterium tuberculosis and Plasmodium falciparum. Mol. Inform. 2015, 34, 292-307. PMid:27490275
View Article PubMed/NCBIFrecer, V.; Miertus, S.; Tossi, A.; Romeo, D. Rational design of inhibitors for drug-resistant HIV-1 aspartic protease mutants. Drug Des. Discov. 1998, 15, 211-231.
Frecer, V.; Burello, E.; Miertus, S. Combinatorial design of nonsymmetrical cyclic urea inhibitors of aspartic protease of HIV-1. Bioorg. Med. Chem. 2005, 13, 5492-5501. PMid:16054372
View Article PubMed/NCBIFrecer, V.; Berti, F.; Benedetti, F.; Miertus, S. Design of peptidomimetic inhibitors of aspartic protease of HIV-1 containing -Phe-Psi-Pro- core and displaying favorable ADME-related properties. J. Mol. Graph. Model. 2008, 27, 376-387. PMid:18678515
View Article PubMed/NCBIDali, B.; Keita, M.; Megnassan, E.; Frecer, V.; Miertus, S. Insight into the selectivity of peptidomimetic inhibitors with modified statine core for plasmepsin II of Plasmodium falciparum over human Cathepsin D. Chem. Biol. Drug Des. 2012, 79, 411-430. PMid:22129033
View Article PubMed/NCBIMegnassan, E.; Keita, M.; Bieri, C.; Esmel, A.; Frecer, V.; Miertus, S. Design of novel dihydroxynaphthoic acid inhibitors of Plasmodium Falciparum Lactate Dehydrogenase. Med. Chem. 2012, 8, 970-984. PMid:22741776
View Article PubMed/NCBIOwono, L.C.; Keita, M.; Megnassan, E.; Frecer, V.; Miertus, S. Design of thymidine analogs targeting thymidilate kinase of Mycobacterium tuberculosis. Tuberc. Res. and Treat. 2013, 2013, 1-13. PMid:23634301
View Article PubMed/NCBIFrecer, V.; Megnassan, E.; Miertus, S. Design and in silico screening of a combinatorial library of antimalarial analogs of triclosan inhibiting Plasmodium falciparum enoyl-acyl carrier protein reductase. Eur. J. Med. Chem. 2009, 44, 3009-3019. PMid:19217192
View Article PubMed/NCBIEsmel, A.; Keita, M.; Megnassan, E.; Beguemsi, T.; Frecer, V.; Miertus, S. Insight into the binding mode of nitrile inhibitors of Plasmodium falciparum falcipain-3, QSAR and pharmacophore models, virtual design of new analogs with favorable pharmacokinetic profiles. SDRP J. Comput. Chem. Mol. Model. 2018, 2, 103-124.
View ArticleDe-Eknamkul, W.; Umehara, K.; Monthakantirat, O.; Toth, R.; Frecer, V.K.; Knapich, L.; Braiuca, P.; Hiroshi, N.; Miertus, S. QSAR study of natural estrogen-like isoflavonoids and diphenolics from Thai medicinal plants. J. Mol. Graph. Model. 2011, 29, 784-794. PMid:21334935
View Article PubMed/NCBIMaple, J.R.; Hwang, M.-J.; Stockfisch, T.P.; Dinur, U.; Waldman, M.; Ewig, C.; Hagler, A. Derivation of class II force fields. I. Methodology and quantum force field for the alkyl functional group and alkane molecules. J. Comput. Chem. 1994, 15, 162-182.
View ArticleGilson, M.K.; Honig, B. The inclusion of electrostatic hydration energies in molecular mechanics calculations. J. Comput. Aid Mol. Des. 1991, 5, 5-20. PMid:2072125
View Article PubMed/NCBIRocchia, W.; Sridharan, S.; Nicholls, A.; Alexov, E.; Chiabrera, A.; Honig, B. Rapid grid-based construction of the molecular surface and the use of induced surface charge to calculate reaction field energies: applications to the molecular systems and geometric objects. J. Comput. Chem. 2002, 23, 128-137. Discovery Studio Molecular Modeling and Simulation Program, version 2.5; Accelrys, Inc.: San Diego, CA, USA, 2009. PMid:11913378
View Article PubMed/NCBIDiscovery Studio Molecular Modeling and Simulation Program, version 2.5; Accelrys, Inc.: San Diego, CA, USA, 2009.
Li, H.; Sutter, J.; Hoffmann, R. Pharmacophore Perception, Development and Use in Drug Design; Güner, O.F., Ed.; International University Line: La Jolla, CA, USA, 2000; pp. 171-189.
QikProp, version 3.7, release 14; X Schrödinger, LLC: New York, NY, USA, 2014.
Insight-II and Discover Molecular Modeling and Simulation Package, version 2005; Accelrys: San Diego, CA, USA, 2005.
N'Guessan, H.; Megnassan, E. In silico design of phosphonic arginine and hydroxamic acid inhibitors of Plasmodium falciparum M17 Leucyl aminopeptidase with favorable pharmacokinetic profile. J. Drug Des. Med. Chem. 2017, 3, 98-125.
View ArticleKoffi, C. K.; Keita, M.; Kre N'Guessan, R.; Luc, C. O.O.; Megnassan, E.; Frecer, V.; Miertus, S. Structure-Based Design and in Silico Screening of Virtual Combinatorial Library of Benzamides Inhibiting 2-trans Enoyl-Acyl Carrier Protein Reductase of Mycobacterium tuberculosis with Favorable Predicted Pharmacokinetic Profiles. Int. J. Mol. Sci. 2019, 20, 4730. PMid:31554227
View Article PubMed/NCBIIssouf, F.; Brice, D.; Frederica, M. K.; Megnassan, E.; Frecer, V.; Miertus, S. Structure-Based Design of Tetrahydroisoquinoline-Based Hydroxamic Acid Derivatives Inhibiting Human Histone Deacetylase 8. J. Comput. Chem. Mol. Model. 2021, 5, 504-538.
View ArticleAvailable Chemicals Directory, Version 95.1, MDL Information Systems, San Leandro, CA. 2003.
Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3-26.
Jorgensen, W.L.; Duffy, E.M. Prediction of drug solubility from monte Carlo simulations. Bioorg. Med. Chem. Let. 2000, 10, 1155-1158. 00172-4
View ArticleJorgensen, W.L.; Duffy, E.M. Prediction of drug solubility from a structure. Adv. Drug Delivery Rev. 2002, 54, 355-366. 00008-X
View ArticleSanjay, K.; Choubey & Jeyaraman, J. Molecular dynamics and quantum chemistry-based approaches to identify isoform-selective HDAC2 inhibitor - a novel target to prevent Alzheimer's disease. J. Recep. Sign. Trans. 2018, 38, 266-278. PMid:29932788
View Article PubMed/NCBIFournier, J.F.; Bhurruth-Alcor, Y.; Musicki, B.; Aubert, J.; Aurelly, M.; Bouix-Peter, C.; Tomas, L. (2018). Squaramides as novel class I and IIb histone deacetylase inhibitors for topical treatment of cutaneous t-cell lymphoma. Bioorg. Med. Chem. Let. 2018, 28, 2985-2992. PMid:30122227
View Article PubMed/NCBI