Dr.Malleshappa N Noolvi
Shree Dhanvantary Pharmacy College · Department of Pharmaceutical Chemistry; Email: noolvimalleshappa@gmail.com
Computational Exploration of New Non-Steroidal Chemo-types as Human Aromatase Inhibitors
Corresponding Author
Affiliation
Dr.Malleshappa N Noolvi*, Mr. Stephen Paul Avvaru, M.Pharm (Ph.D.)1, Dr. Ashutosh Dash, Ph.D.2, Dr. Sudipta Chakraborty, Ph.D3, Dr. Tejraj M Aminabhavi, Ph.D4, Uttam A. More, Ph.D.5
1. Department of Pharmaceutical Chemistry, Shree Dhanvantary Pharmacy College, Kim, Surat 394110, India.
Phone No: 7730006666, Fax: +91 2621 233344, E-Mail: stephen.avvaru@gmail.com
2. Head, Radiopharmaceuticals Division, Bhabha Atomic Research Centre, Mumbai – 400085, India.
Phone No: 91-22-25595372, Fax no: 912225505151; E-Mail: adash@barc.gov.in
3. Radiopharmaceuticals Division, Bhabha Atomic Research Centre, Mumbai – 400085, India.
Phone No: 9122255903909/0626/0627, Fax no: 912225505151; E-Mail: sudipta@barc.gov.in
4. Chairperson Advisory Committee, Shree Dhanvantary Pharmacy College, Kim, Surat-394110, India.
Phone No: 09449821279, Fax: +91 2621 233344; E-Mail:aminabhavi@yahoo.com
5. Department of Pharmaceutical Chemistry, Shree Dhanvantary Pharmacy College, Kim, Surat-394110, India.
Phone No: 8401841577, Fax: +91 2621 233344; E-mail: uttamsvd@gmail.com
Article Reviewed By:
Xiaoguang Xie(xgxie@ynu.edu.cn)
Mohamed AbdElAziz Khamis(mohamed.khamis@ejust.edu.eg)
Zhi-Yong Tian(tzynew@163.com)
Petko Alov(petko@biophys.bas.bg)
Citation
Dr.Malleshappa N. Noolvi,
Abstract
The development of the third-generation aromatase inhibitors viz. Letrozole, Anastrozole and Exemestane as first line drugs for estrogen dependent breast cancer treatment is a momentous achievement. The inevitability of prolonged use of these first line drugs to treat breast cancer, develops different resistance mechanisms in the cancer cell. The quest for new class of aromatase inhibitors is essential to overcome the plausible resistance and unwanted side effects due to chronic therapy. Considering the magnitude for the necessity of newer generation aromatase inhibitors, the present research proposal aims at the design of new series of heterocyclic rings like Benzothiazole, Benzimidazole 1,3,4-thiadiazole and 1,3,4-oxadiazole, imidazo[2,1-B][1,3,4]thiadiazole derivatives as aromatase inhibitors using Ligand Based 3D QSAR Pharmacophore Modeling and Molecular docking studies. A 3-point pharmacophore with one hydrogen bond acceptor (A) and two aromatic rings (R) was selected as pharmacophore hypothesis for building the 3D QSAR model. Training set correlation with Partial Least Square factors (R2=0.99, SD=0.1265, F=470.6, P=1.033e-018) and the test set correlation (Q2 = 0.7854, RMSE = 0.5284, Pearson R = 0.9111) N = 34 was chosen among the four different PLS factors generated. Letrozole and vorozole were also included in the training and test sets for the reference. The pharmacophore-based alignment hypothesis was done for the new series of ligands to predict the activity. Compounds 35, 40 and 50 (predicted activity 0.8697, 0.737991 and 0.865794 respectively) have shown positive findings with respect to Letrozole and Vorozole. The Docking results also gave supportive evidence for the ligand 50 with good docking score. Interestingly the S isomers had given better docking scores when compared R counterparts. Methionine 374 H-bond interaction with ligand is needed for the selectivity to aromatase active site, some of the proposed ligands also shown H-bond interaction with Threonine 310, Alanine 306 amino acid residues. This alternate binding interactions at the opening of the active site of aromatase may open the new class of aromatase inhibitors.
Introduction
Aromatase is the enzyme which is predominantly present in woman [1]. It selectively converts androgens specifically C19 steroids with 4-ene-3-one system to estrogens by aromatizing the ring A of steroids. Estrogens in woman plays an important role in reproduction and other homeostasis functions of cell. In premenopausal women Gonadotropin releasing hormone and Follicle stimulating hormone helps in monitoring the estrogen production by negative feedback mechanism. However in postmenopausal women there is no such mechanism. This may result in imbalance in homeostasis of cell that causes breast cancer [2], Endometriosis, Lung cancer, Hepatic cancer and other pathological conditions [3]. Around 70% of postmenopausal breast cancer patients are found to be estrogen-dependent breast cancer cases [4]. Inhibiting the aromatase enzyme is one of the important approach to treat the estrogen dependent breast cancer [5]. The Aromatase inhibitors (AIs) used as first line drug in the treatment of postmenopausal breast cancer women are Anastrazole, Exemestane, Letrozole. These drugs are also approved for early and advanced cases of breast cancer in postmenopausal women [6,7]. These AIs bind the aromatase and prevent the binding of natural substrate (androgens).
Aromatase inhibitors chronic monotherapy or their combination with other targeted agents reported developing resistance along with side effects like osteoporosis, cardiovascular complications and others [7,8]. This resistance developed by breast cancer cells against aromatase inhibitors implicates significant reasons like integral tumor insensitivity to estrogen, aromatase inhibition unresponsiveness, production of estrogenic hormones independent of aromatase, triggering of non-endocrinal signaling pathways, bypassing apoptosis pathway and selection of hormone-insensitive cellular clones during treatment [2,9,10]. Hence aromatase inhibitors resistance has been confirmed as the major obstacle to optimal therapy management.
In-silico studies are one of the effective tools to develop new drug prototypes in short period of time to contest with the pace of new drug discovery demand. Several researchers have done insilico studies using Pharmacophore modeling [11], Structure-Guided Design using crystal structure of human placental aromatase, High-Throughput Docking which facilitated the yield of promising novel AIs and insights to the aromatase active site [12,13,14,15]. The advanced models like membrane-bound all-atom molecular dynamics simulations of aromatase reveal new insights into the fluctuations of the active site, the access channel, heme-proximal cavity, and dynamic quaternary organization of ER membrane-embedded aromatase. [16,17,18].
Some of the findings reveal that the polar, aromatic and non-polar residues of aromatase active site play a vital role in interacting with AIs [4]. The presence of heme prosthetic group at the active site provides the electrons in the form of Ferric peroxide for the conversion of androgens to estrogens [19]. Park et al with the molecular simulations studies of the aromatase found that ASP-309 residue present at the active site of aromatase is critical in active site access channels [18]. Another key interaction to be mentioned is the presence of a basic nitrogen atom (imidazole or triazole) in the structure of aromatase inhibitors apically Co-ordinate the iron atom of the heme prosthetic group of the aromatase enzyme [1,20].
The present research aims at the design of new series of heterocyclic rings like Benzothiazole, Benzimidazole 1,3,4-thiadiazole and 1,3,4-oxadiazole, imidazo[2,1-B][1,3,4]thiadiazole derivatives as aromatase inhibitors using Ligand Based 3D QSAR Pharmacophore Modeling and Molecular docking studies. The pharmacophore modeling was carried out using pharmacophore alignment and scoring engine (Phase). The Phase generates partial least-square (PLS) regression, which gives significant prediction model. The predicted activity of the newly designed ligand was done using the 3D QSAR pharmacophore model.The molecular docking of the newly designed ligands was performed using Maestro.
Materials & Methods
All computational and molecular modeling studies were carried out using the software Schrodinger, LLC, USA, 2009.
2.1. Pharmacophore based 3D-QSAR Method
2.1.1. Dataset
Thirty-four molecules of substituted letrozole based analogs as aromatase inhibitors were chosen from research work of Doiron J et al. and used for QSAR analysis [21]. All the IC50 (µM) values had been obtained using the same assay method (using the P450 Inhibition Kit CYP19/MFC (BD Biosciences, Two Oak Park Bedford, MA, USA). The IC50 values of reference compounds were checked to ensure that no difference occurred between different groups. These Letrozole based analogs series showed wide variations in their structures and potency profiles with IC50 (µM) ranging from 0.002 to 49.98. The 2D structures of 34 molecules of substituted letrozol analogs (training and test), 31 newely designed ligands were drawn using ChemBioDraw Ultra version 12.0, 2010. The 3D conformers and minimization of ligands were generated using “LigPrep” incorporated in PHASE.
2.1.2. Common phramacophore hypothesis generation:
The common pharmacophores hypothesis (CPH) were developed using Phase, version 3.1, Schrödinger, LLC, USA, 2009. The pharmacophore features were defined and identified from the built in set of common pharmacophore features in the PHASE namely hydrogen bond acceptor (A), hydrogen bond donor (D), hydrophobic group (H), negatively changed group (N), positively charged group (P), aromatic ring (R). From the generated variants, one Hydrogen bond acceptor and two Aromatic rings (ARR) were inferred based on the highly active molecule (Letrozole). These CPH were examined by scoring function to get the best alignment of the ligands which groups together similar pharmacophores according to their inter-site distances (fig.1). The quality of each alignment is measured by alignment score (RMSD in the site-point positions), Vector score (average cosine of the angles formed by corresponding pairs of vector features, ARR in the aligned structures) and volume score (overlap of van der Waals models of the non-hydrogen atoms in each pair of structures).
The alignment was measured using survival score, defined as: S=WsiteSsite + Wvec Svec + Wvol Svol + Wsel Ssel + Wmrew, where the W’s are the weights and the S’s the scores; Ssite represents alignment score, Svec represents vector score, Svol represents volume score and Ssel represents selectivity score. Wsel has default value of 0.0 and Wsite, Wvec, Wvol, Wrew, has default values of 1.0 which are used for the hypothesis generation. Wmrew, represents reward weights defined by m-1, where m is the number of actives that match the hypothesis.
2.1.3. Generating QSAR model
In order to generate a validated QSAR model for the resolve of meaningful prediction, the available data set was randomly divided into a training set of 24 molecules and a test set of 10 molecules by incorporating IC50 activity as dependent variable and chemical diversity of aligned training set ligands. The regression was done by a partial least squares (PLS) method, in which a series of models were constructed with four different PLS factors. The PLS factor 4 (# Factor) was chosen (table 1), because of the Training set correlation with Partial Least Square factors (R2=0.99, SD=0.1265, F=470.6, P=1.033e-018) gave the best overall significance of model and statistical significance. The parameters were used to evaluate the test set prediction correlation (Q2 = 0.7854, RMSE = 0.5284, Pearson R = 0.9111). F is the variance ratio. Large values of F indicate a more statistically significant regression. P is significance level of variance ratio. Smaller values of P indicated a greater degree of confidence. Q-squared is the value of Q2 for the predicted activities. Pearson-R is the value for the correlation between the predicted and observed activity for the test set. The 3D QSAR models that met all these criteria concurrently gave the best predictive power.
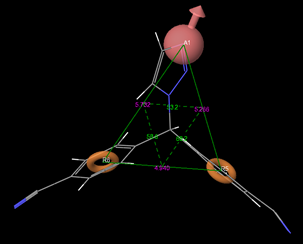
Fig 1: Geometry of pharmacophore hypothesis
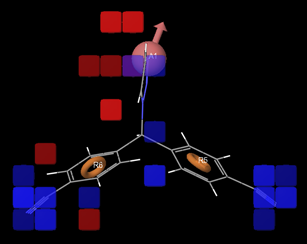
Fig 2: positive and negative co-efficient of ligand in 3D QSAR model
The generated results like predicted activity of the training and test molecules were evaluated. All the compounds along with their activity and predicted activities are presented in Table 2.
The graph of Phase predicted activity and Phase activity was generated for both training and test molecules. The IC50 values in the graph are represented with polychromatic markings to correlate with phase predicted activity and phase activity. Higher values (positive) on the scattered plot indicates the higher potency of the ligands. (Fig. 3 & 4).
The equations for Partial Least Square Regression analysis for training molecules were derived from the following parameters.
m is the number of PLS factors in the model
n is the number of molecules in training set
df1 = m+1 is the degrees of freedom in model
df2 = n- m-2 is the degrees of freedom in data
yi is observed activity for training set molecule i
ŷi is predicted activity for training set molecule i

The statistical quantities describing the test set predictions are described below
T is test set of molecules
nT is number of molecules in T
yj is observed activity for molecule j ∈ T
ŷj is predicted activity for molecule j ∈ T

Fig 3: 2D structures of ligands used in 3D QSAR
Fig 4: 2D structures of ligands used in 3D QSAR
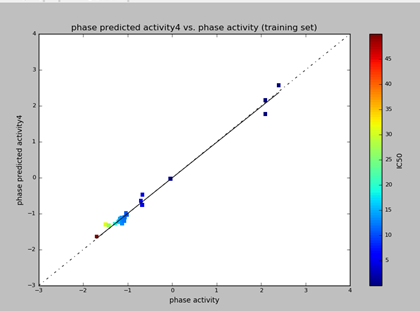
Fig 5: The scattered plot of phase predicted activity and phase activity of training set
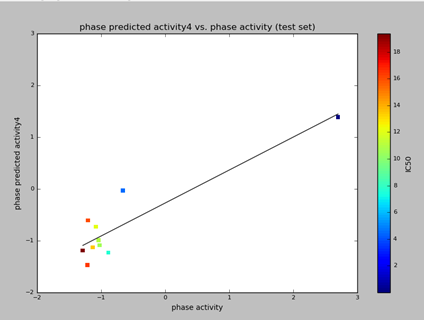
Fig 6: The scattered plot of phase predicted activity and phase activity of test molecules
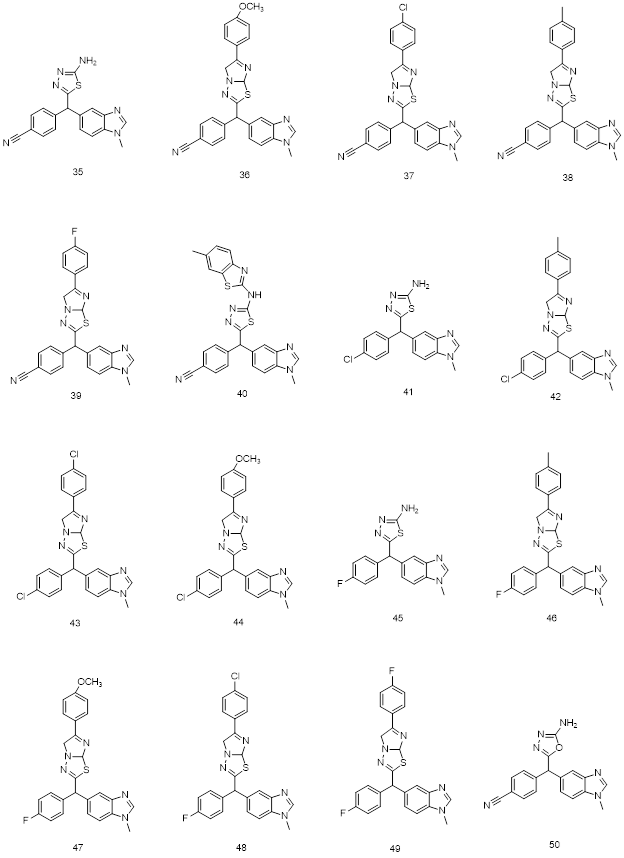
Fig.7: 2D structures of new chemo-types used in 3D QSAR and Molecular docking
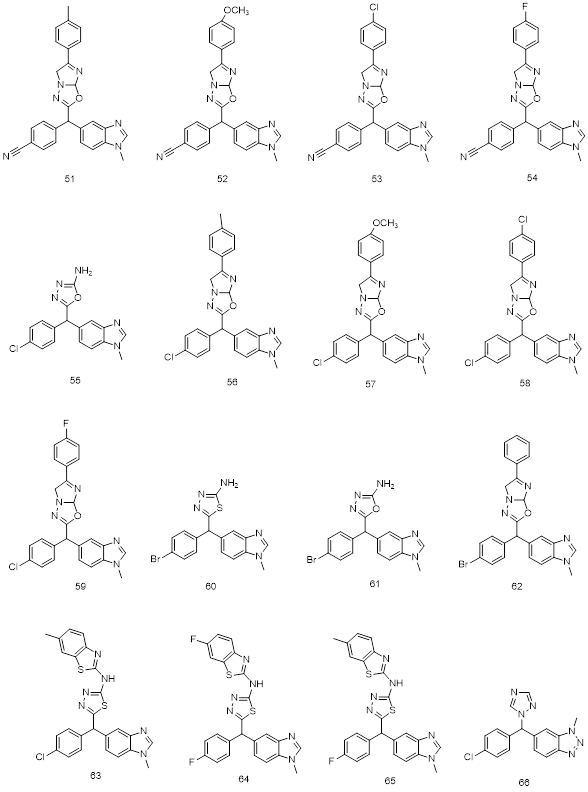
Fig.8: 2D structures of new chemo-types used in 3D QSAR and Molecular docking
2.2. Docking
Taking advantage of the unexplored chemo-types as aromatase inhibitors and generated hypothesis data of ligands from the QSAR results, new subclass of ligands were proposed. Docking studies were performed using Aromatase enzyme (PDB:3S7S) on maestro-Schrodinger 9.1. 34 molecules including Letrozole and Vorozole were subjected to docking on x-ray crystallographic structure of aromatase (PDB code: 3S7S). This involved stepwise functions like protein selection, protein preparation, grid generation, ligand preparation and ligand docking studies. Ligads with only good comparable docking scores are presented in table 5 as well as in Figures 5 & 6.
All the ligands structures were drawn using ChemBioDraw Ultra 12.0. The ligands were then subjected to ligprep for generating 3D optimal and minimum energy conformers. The aromatase protein having Exemestane as a substrate in its active site (PDB: 3S7S from RCSB Protein Data Bank) was selected for the docking studies. Once the protein was imported on to the maestro docking panel, the source ligand (Exemestane) in the active site was removed followed by the preprocess like optimization and minimization of energies removal water molecules etc. Then a grid is generated at the active site of aromatase protein (PDB code: 3S7S). The prepared new set of ligands were then subjected to glide for the ligand docking. The glide docked poses were minimized by local optimization features from prime. OPLS_2005 force field and GB/SA continuum solvent model were used for calculating the energies of ligand docking complexes. The scoring algorithm was then carried on energy minimized poses to generate Glide/Dock score. The each new ligand docked poses on the Aromatase active site displayed on the workspace were enhanced by using Glide XP Visualizer to display representations like important hydrophobic, pi-pi stacking interactions and hydrogen bonds between the receptor and the ligand in the Workspace.
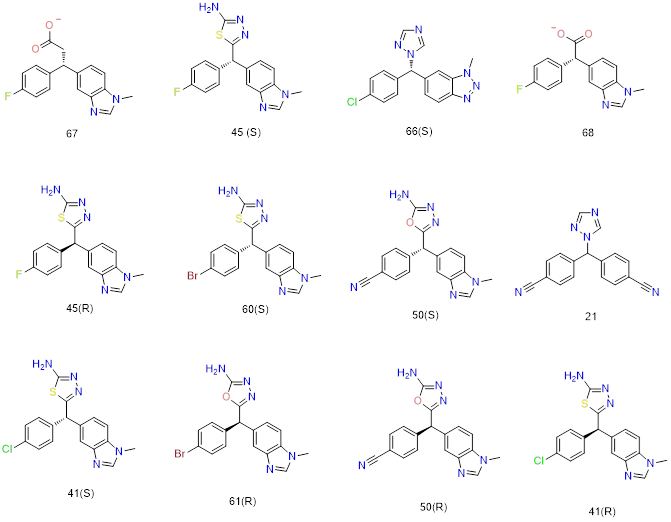
Fig.9: selected 2D structures among the several docked ligands used in molecular docking.
Results
3.1. QSAR results
3-point pharmacophore model ARR, with one hydrogen bond acceptor (A), and Two aromatic rings (R). The generated pharmacophore-based alignment hypothesis (Q2 = 0.785, RMSE = 0.528, Pearson R = 0.911, N = 34) was used to derive a predictive activity of 3D-QSAR model for the training set and test set.
The visualized 3DQSAR model in the Workspace (Fig 2) is used to identify ligand features that contribute positively or negatively to the predicted activity. Letrozole is represented in this as prototype 3D QSAR model (Fig.2). The region of blue cubes at Cyano functional group of the ligand indicates the favorable environment for electron withdrawing groups which affects the biological activity positively. The blue cubes on hydrogen bond acceptor region also indicate favorable environment to attach hydrogen bond acceptor atoms like Oxygen and Nitrogen which affects biological activity positively. The red cubes slightly away from the H-bond acceptor region indicate unfavorable environment to attach any functional groups which affects biological activity negatively. The representation of higher vector score values indicates prominent role of the vector features like acceptors, aromatic rings in the aligned structures. The volume score is based on the overlap of van der Waals models of the non-hydrogen atoms in each pair of structures. Thus the hydrogen bond acceptor (A) like oxygen or Nitrogen is essential for increasing the potency of the ligand (ligands 35 & 50). Attaching any hydrophobic functional groups near hydrogen bond acceptor region (A) has resulted in negative predicted activity values (Table 3). The extension of hydrogen bond acceptor (A) region with imidazo[2,1-B][1,3,4]thiadiazole or imidazo[2,1-B][1,3,4]oxadiazole ring resulted in decreased potency of the ligand which can be seen with the negative predicted activity values. However attachment of benzothiazole (aromatic) in this region influenced the biological activity positively (ligand 40) because of the degree of free rotation of bond on the nitrogen unlike fused heterocyclic rings.
The proposed molecules in the 3D-QSAR results showed that ligands 35, 40 and 50 have good predicted activity (0.8697, 0.7379 and 0.86579 respectively) close to Vorozole. Letrozole showed far higher predicted activity (1.602764) as expected.
3.2. Docking results
Using Glide XP Visualizer on the maestro workspace, the ligand receptor interactions were enhanced by developing the active site surface mesh. One of the interesting finding is the aromatase receptor site shape. It looks typically like an Iodine flask. The L- phenylalanine 221, L-Valine 313 makes up the hydrophobic conical entrance to the active cite. L-Threonine 310, L-Serine 478 (negatively charged amino acids in red) forms the neck region and L-Aspartic acid 309 acts as a lid, allowing or preventing the substrate entering to the active cite. Methionine 374, L-Alanine 306, Valine 369, Valine 370, L-Leucine 372, Valine 373, L-Tryptophan 224, Phenylalanine 134, L-Leucine 477 forms the hydrophobic conical body (amino acids in green). Heme prosthatic group (grey) forms the flat bottom of the flask which plays the critical role in providing electrons to the substrate for aromatization.
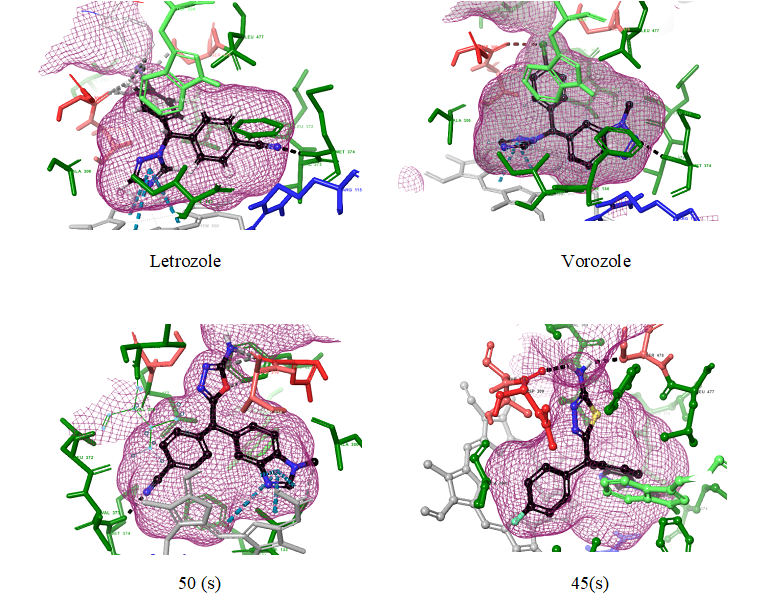
Fig10: 3D representation of Ligand receptor interactions
All the ligands that fit the active site with good docking score gave the efficiency of the ligand. However other parameters like H bond interaction and interaction of the ligand with heme prosthetic group present at the active site confers the selectivity of ligand to aromatase. Hence the generated docking score was then interpreted using H-bond interactions and hetero atoms interaction with heme prosthetic group of aromatase active site, and other hydrophobic interactions with reference to the standard drugs Letrozole and Vorozole.
Ligands 67 and 45(s) (fig 10) represented better docking score than Letrozole and Vorozole. The H-Bond interactions of 67 and 45(s) with the amino acids at the active site varied with that of Letrozole and vorozole interactions. Letrozole and Vorozole Characteristically had H-bond interaction with MET 374 amino acid of aromatase. Heme prosthetic group of aromatase involves in most of the Pi-Pi stacking interaction with 5 membered heterocyclic aromatic functional group (like triazole ring in Letrozole) of the ligands [1,20]. The H-bond interaction of electron withdrawing group (like -C≡N in letrozole), with MET 374 or THR 310 is important for selectivity of the drug to active site [1,4,22,].
We found that S configuration of all ligands showed better docking scores compared to their R counterparts (Table 4). This indicates the S configuration of the ligands are mostly exposed to important binding interactions, that is MET 374 and Heme prosthatic group in the active site. Along with these the presence of Phenyl/aromatic functional group of ligands played an important role in hydrophobic interactions at the active site of aromatase. The docking results of R-configuration ligands presented that ARG 115, PHE 134, TRP 224 as the other important binding interactions of aromatase which are involved in a hydrophobic or Pi-Pi stacking interaction with Phenyl or aromatic functional group of the ligands.
The ligands which have H-bond interaction with an amino acid like MET 374, THR 310 also have Pi-Pi stacking interaction with heme prosthatic group as it can be observed in ligand 45(s) and 50(s). The presence of sixteen different aromatase mutants at amino acid residues Isoleucine133, Phenylalanine235, Isoleucine395, Isoleucine474, Glutamic acid 302, Proline 308, Aspartic acid 309, Threonine310, Serine 478, and Histidine 480 was reported [23, 24]. These mutants dramatically increases or decreases the binding affinity of ligand with the amino acid residues at the active site of aromatase [22]. From the docking results of the research, we found that ligands like 45 (s) which have primary amine functional group (-NH2) attached to 5 membered heterocyclic ring have H-bond interaction with THR 310 and SER 478. This dual interaction increases the possibility of alternate binding interactions when the amino acid residues are mutated.
Conclusion
In the present study, ligand based 3D QSAR pharmacophore modeling and docking studies of non-steroidal Aromatase Inhibitors was done for a series of novel Benzothiazole, Benzimidazole, 1,3,4-thiadiazole, 1,3,4-oxadiazole, imidazo[2,1-B][1,3,4]thiadiazole derivatives. 3D Pharmacophore model ARR.1 was chosen with PLS factor 4 as the best model because the Training set correlation with Partial Least Square factors gave the best overall significance of model and statistical significance and the test set prediction correlations. The aromatase inhibitor potency of proposed ligands was influenced by Hydrogen bond acceptor and electron withdrawing groups on aromatic rings which is represented in the vector score and on the 3D pharmacophore model (fig.3). Benzothiazole, 1,3,4-thiadiazole, 1,3,4-oxadiazole containing n-methyl benzimidazole derivatives exhibited good predicted activities. However imidazo[2,1-B][1,3,4]thiadiazole derivatives gave negative predicted activity values. Any fused heterocyclic ring on the hydrogen bond acceptor hindered the aromatase inhibitory potency. The most virtually potent compounds of proposed ligands are 35, 40 and 50.
The molecular docking studies were carried out on 34 molecules including Letrozole and Vorozole. The molecules with good docking score were chosen here. The activity of the molecules was analyzed based on the docking score and hydrogen bond interaction of the ligand with the receptor. The H-bond interaction of ligands with Methionine 374 represents the selectivity of ligands with the aromatase active site. The proposed ligands binding with different amino acids of receptor and differentially binding with heme prosthetic group of aromatase active site emphasizes not only selectivity but also the alternate binding interactions at the active site of aromatase when compared with Letrozole and vorozole. These alternate binding interactions may help overcome mutated amino acids irresponsiveness due to chronic therapy. These novel chemo-types could be useful in developing rationale non-steroidal molecules for the aromatase inhibitory activity.
Acknowledgement
The authors would like to thank the Member Secretary, Board of Research in Nuclear Science (BRNS), Government of India, for funding the Project (Sanction No.35/14/25/2017-BRNS/35240) and Dr. N. D. Jivani, President, Shree Dhanvantary Pharmacy College for providing necessary facilities.
References
Ghosh D, Griswold J, Erman M, Pangborn W. Structural basis for androgen specificity and oestrogen synthesis in human aromatase .Nature. 2009; 457:219-223 PMid:19129847
View Article PubMed/NCBIChumsri S, Howes T, Bao T, Sabnis G, Brodie A; Aromatase, Aromatase Inhibitors, and Breast Cancer; J. Steroid Biochem Mol. Biol. 2011, 125(1-2): 13–22. PMid:21335088
View Article PubMed/NCBIKim J.J, Kurita T, Bulun S.E; Progesterone action in endometrial cancer, endometriosis, uterine fibroids and breast cancer; Endocr Rev. 2013; 34(1):130-162 PMid:23303565
View Article PubMed/NCBISuvannang N, Nantasenamat C, Isarankura-Na-Ayudhya C, Prachayasittikul V, Molecular Docking of Aromatase Inhibitors; Molecules 2011, 16, 3597-3617.
View ArticleBrodie A.M, Schwarzel W.C, Shaikh A.A, Brodies H.J; he effect of an aromatase inhibitor, 4-hydroxy-4-androstene-3,17-dione on estrogen dependent process in reproduction and breast cancer. Endocrinology; 1977, 100, 1684-95. PMid:404132
View Article PubMed/NCBILønning P.E, Eikesdal H.P, Aromatase inhibition 2013: clinical state of the art and questions that remain to be solved; Endocr Relat Cancer; 2013, 20(4), R183–R201. PMid:23625614
View Article PubMed/NCBIMcArthur H.L, Morris P.G; Aromatase inhibitor strategies in metastatic breast cancer; Int J Womens Health. 2009; 1, 67–72.
View ArticleMa CX, Reinert T, Chmielewska I, Ellis MJ, Mechanisms of aromatase inhibitor resistance; Nature Reviews Cancer 2015 15: 261–275 PMid:25907219
View Article PubMed/NCBIMiller W.R, Larionov A.A; Understanding the mechanisms of aromatase inhibitor resistance; Breast Cancer Research; 2012, 14, 201. PMid:22277572
View Article PubMed/NCBIBrodie A, .Sabnis G, Jelovac D. Aromatase and breast cancer; J Steroid Biochem Mol Biol. 2006; 102(1-5): 97–102. PMid:17113978
View Article PubMed/NCBISchuster D, Laggner C, Steindl T.M, Palusczak A, Hartmann R.W, Langer T; Pharmacophore modeling and in silico screening for new P450 19 (aromatase) inhibitors, J. Chem. Inf. Model. 2006, 46(3), 1301-11. PMid:16711749
View Article PubMed/NCBIGhosh D, Lo J, Morton D, Valette D, Xi J, Griswold J, Hubbell S, Egbuta C, Jiang W, An J, Davies H.M.L; Novel Aromatase Inhibitors by Structure-Guided Design; J. Med. Chem., 2012, 55 (19), 8464–76 PMid:22951074
View Article PubMed/NCBICaporuscio F, Rastelli G, Imbriano C, Rio A.D; Structure-Based Design of Potent Aromatase Inhibitors by High-Throughput Docking; J. Med. Chem., 2011, 54 (12), 4006–17 PMid:21604760
View Article PubMed/NCBIRoy P.P., Roy K.; Docking and 3D-QSAR studies of diverse classes of human aromatase (CYP19) inhibitors. J Mol Model., 2010, 16,1597–1616. PMid:20195667
View Article PubMed/NCBIDai Y., Wang Q., Zhang X., Jia S., Zheng H., Feng D., Yu P. Molecular docking and QSAR study on steroidal compounds as aromatase inhibitors. Eur J Med Chem., 2010, 45, 5612–5620. PMid:20926163
View Article PubMed/NCBILo J, Nardo G.D, Griswold J, Egbuta C,Jiang W, Gilardi G, Ghosh D, Structural Basis for the Functional Roles of Critical Residues in Human Cytochrome P450 Aromatase, Biochemistry, 2013, 52 (34), 5821–9 PMid:23899247
View Article PubMed/NCBIJiang W, Ghosh, D; Motion and flexibility in human cytochrome p450 aromatase; PloS one, 2012, 7(2), e32565 PMid:22384274
View Article PubMed/NCBIJiho P, Luke C, Amaro R.E; Molecular Simulations of Aromatase Reveal New Insights Into the Mechanism of Ligand Binding; J. Chem. Inf. Model. 2013, 53, 2047-56 PMid:23927370
View Article PubMed/NCBIYoshimoto F.K., Guengerich F.P. Mechanism of the third oxidative step in the conversion of androgens to estrogens by cytochrome P450 19A1 steroid aromatase. J.Am.Chem. Soc., 2014, 136, 15016-15025. PMid:25252141
View Article PubMed/NCBIRecanatini M., Cavalli A., Valenti P. Nonsteroidal aromatase inhibitors: recent advances. Med. Res. Rev., 2002, 22, 282−304 PMid:11933021
View Article PubMed/NCBIDoiron J, Soultan A.H, Richard R, Touré M.M, Picot N, Richard R, Cuperlović-Culf M, Robichaud G.A, Touaibia M; Synthesis and structure-activity relationship of 1- and 2-substituted-1,2,3-triazole letrozole-based analogues as aromatase inhibitors; Eur. J. Med. Chem. 2011, 46(9), 4010-24. PMid:21703734
View Article PubMed/NCBIHong Y., Li H., Yuan Y., Chen S; Molecular characterization of aromatase, Ann N Y Acad Sci. 2009, 1155, 112–120. PMid:19250198
View Article PubMed/NCBIKao Y.C, Cam L.L, Laughton CA, Zhou D, Chen S; Binding characteristics of seven inhibitors of human aromatase: a site-directed mutagenesis study. Cancer Res. 1996, 56(15), 3451-60. PMid:8758911
PubMed/NCBIKao Y.C, Korzekwa K.R, Laughton C.A, Chen S; Evaluation of the mechanism of aromatase cytochrome P450. A site-directed mutagenesis study. Eur J Biochem. 2001, 268(2), 243-51. PMid:11168357
View Article PubMed/NCBI