Daniel Glossman-Mitnik
Email: daniel.glossman@cimav.edu.mx
© 2019 Sift Desk Journals. All Rights Reserved
VOLUME: 3 ISSUE: 1
Page No: 235-251
Daniel Glossman-Mitnik
Email: daniel.glossman@cimav.edu.mx
Juan Frau1y and Daniel Glossman-Mitnik1,2*y
1Departament de Qu mica, Universitat de les Illes Balears, Carretera a Valldemossa, Km 7.5, 07122 Palma de Mallorca, Spain.
2Laboratorio Virtual NANOCOSMOS, Departamento de Medio Ambiente y Energ a, Centro de Investigacion en Materiales Avanzados, Miguel de Cervantes 120, Complejo Industrial Chihuahua, Chih 31136 Chihuahua, Mexico.
Mirza Wasif Baig(wasifbaig.mirza@jh-inst.cas.cz)
Goar S%c3%a1nchez-Sanz(goar.sanchez@ichec.ie)
A Pawlukoj%c4%87(a.pawlukojc@ichtj.waw.pl)
Ramiro F Quijano-Qui%c3%b1ones(ramiro.quijano@uady.mx)
Daniel Glossman-Mitnik, Computed Local Chemical Reactivity of Melanoidins Red M1 and Red M2 Using Conceptual DFT(2019)SDRP Journal of Computational Chemistry & Molecular Modelling 3(1)p:235-251
This study assessed ten density functionals, including CAM-B3LYP, LC-ωPBE, M11, M11L, MN12L, MN12SX, N12, N12SX, ωB97X, and ωB97XD related to the Def2TZVP basis set together with the SMD solvation model in the calculation of the molecular properties and structures of the Red-M1 and Red-M2 intermediate melanoidin pigments. The global and local chemical reactivity descriptors for the systems were calculated via the Conceptual Density Functional Theory. The choice of the active sites applicable to nucleophilic, electrophilic as well as radical attacks was made by linking them with the Fukui function indices, the electrophilic Parr functions, and the condensed dual descriptor ∆f(r) over the atomic sites. The prediction of the maximum absorption wavelength directly from the HOMO–LUMO tended to be considerably accurate relative to the experimental values for the MN12SX and N12SX density functionals. This study found the MN12SX and N12SX density functionals to be the most appropriate to predict the chemical reactivity of the molecules under consideration.
Keywords: Melanoidins; Red-M1; Red-M2; Conceptual DFT; Chemical Reactivity; Dual Descriptor; Parr Function; Maximum Absorption Wavelength
Melanoidins are brown pigments formed during the reaction of reducing sugars with amino groups, derived from amino acids, amines, and proteins. Their chemical structure is largely unknown, but their functional properties have been a matter of intensive study because of their abundance in heat-treated foods. Melanoidin formation contributes to the development of aroma and color in processed foods, as well as food texture and beverage viscosity [1].
Melanoidins are considered as the final products of a process known as non-enzymatic browning or the Maillard reaction. A similar reaction between reducing sugars and biological molecules takes place under physiological conditions, in a process usually called glycation. Glycation has a significant impact on the aging of living organisms and the pathology of some diseases [1]. We have recently dedicated much effort to understand the glycation process, which is mainly associated with certain diseases like Diabetes, Alzheimer, and Parkinson, as well as the chemical reactivity of the reducing carbohydrates, amino acids, and small peptides that participate in it [2–9].
Although color formation is the primary characteristic of the Maillard reaction, our knowledge of the colored moieties responsible for the coloration is still only rudimentary. The low-weight intermediate colored products are known as colored Maillard reaction products (CMRP) and the color produced can be readily measured by reading the absorbance in the visible region of the spectrum, with the typical wavelengths used being 360 and 420 nm [10].
These CMRPs are of great interest not only in the food industry, but also for their potential application as photosensitizers, as antioxidants related to health and as colorants to be used in dye-sensitized solar cells for the production of alternative energy. Thus, there is a great interest in studying their chemical properties, with special emphasis on their molecular reactivity.
Conceptual Density Functional Theory (CDFT, also known as Chemical Reactivity Theory) is a powerful tool for the prediction, analysis, and interpretation of the outcome of chemical reactions [11–14].
Two interesting CMRPs amenable to being studied through conceptual DFT are Red-M1 and its diastereomer Red-M2 [15], which are red pigments isolated from the reaction system between D-xylose and glycine. Therefore, we believe that it could be of interest to apply the concepts of DFT to the study of the chemical reactivity of these red pigments.
In order to obtain quantitative values of the conceptual DFT descriptors, it is necessary to utilize the Kohn–Sham (KS) theory through calculations of the molecular density, the energy of the system, and the orbital energies - specifically those related to the frontier orbitals; that is, the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) [16–21].
In recent years, there has been significant interest in the use of range-separated (RS) exchange-correlation functionals in KS DFT [22]. These functionals partition the 1/r12 operator in the exchange term into short- and long-range components, with a range-separation parameter, ω, controlling the rate at which the long-range behavior is attained. The value of ω can be fixed or it can be “tuned” on a system-by-system basis by minimizing some tuning norm. The optimal tuning approach is based on the knowledge that in exact KS and generalized KS theory, the energy of the HOMO, εH(N), for an N-electron system should be exactly −IP(N), where IP is the vertical ionization potential calculated with a given functional as the energy difference E(N−1) − E(N). With approximate functionals, the differences between εH (N) and −IP(N) can be very large. Optimal tuning implies a non-empirical determination of a system-specific range-separation parameter ω in an RSE functional and, optionally, other parameters, such that εH(N) = −IP(N) is satisfied to the best possible degree [23]. Although there is no equivalency to this prescription for the electron affinity (EA), and the LUMO of the neutral species, it can be said that εH(N+1) = −EA(N), thus making it possible to find an optimized ω value that is simultaneously optimized for getting both properties, rendering it useful for an easy prediction of conceptual DFT descriptors. In previous works [2–9], we have termed this simultaneous prescription as the “KID procedure” for its analogy with the Koopmans’ theorem.
This means that the goodness of a given density functional for the purpose of predicting the conceptual DFT descriptors directly from the properties of the neutral molecule can be estimated by checking how well it follows the KID procedure. However, this tune optimization is system-dependent, and it must be performed for each molecule. Thus, it will be interesting to consider several recent density functionals that have shown great accuracy across a broad spectrum of databases in chemistry and physics and where the value of ω is fixed to evaluate their performance in fulfilling this practical technique.
The objective of this work is to conduct a comparative study of the performance of some recent density functionals for reproducing the chemical reactivity descriptors of the Red-M1 and Red-M2 pigments within the KID formalism to gain insight into their molecular properties that can be used for future studies on the chemical reactivity of colored melanoidins of larger molecular weights formed in the reaction of reducing sugars with peptides and proteins.
Theoretical Background
As this work is part of an ongoing project, the theoretical background is similar to that presented in previous research [2–9] and will be shown here for the sake of completeness. The chemical potential μ is defined within the conceptual framework of DFT [12,24] as:
μ=(∂E/∂N)(v(r))= -χ (1)
where χ is the total electronegativity. In turn, the global hardness η is defined as:
η= ((∂2 E)/(∂N2 ))(v(r)) (2)
If we consider the KID procedure mentioned in the introduction together with a finite difference approximation, then the above expressions can be written as:
μ=-1/2 (I+A)≈1/2 (εH+εL) (3)
η=(I-A)≈(εL-εH) (4)
where εH and εL are the energies of HOMO and LUMO, respectively.
The electrophilicity index ω has been defined as [25]:
ω=μ2/2η=〖(I+A)〗2/(8(I-A))≈(εL+εH)2/8(εL-εH) (5)
Applying the same ideas, two definitions of Fukui functions depending on total electronic densities are obtained:
f+ (r)= ρ(N+1) (r)-ρN (r) (6)
f- (r)= ρN (r)-ρ(N-1) (r) (7)
where ρ(N+1) (r) , ρN (r) and ρ(N-1) (r) are the electronic densities at point r for the system with N1, N, and N1 electrons, respectively. The first function, , has been associated to reactivity for a nucleophilic attack, and thus it measures the intramolecular reactivity at the site r towards a nucleophilic reagent. The second, , has been associated to reactivity for an electrophilic attack, and thus this function measures the intramolecular reactivity at the site r toward an electrophilic reagent [24].
Morell et al. [13,26–31] proposed a local reactivity descriptor (LRD), which is called the dual descriptor (DD) f(2) (r)≡∆f(r):
∆f(r)= (∂f(r)/∂N)v(r) (8)
This can be condensed over the atomic sites: when ∆fk > 0, the process is driven by a nucleophilic attack on atom k, and then the atom acts as an electrophilic species; conversely, when ∆fk < 0, the process is driven by an electrophilic attack over atom k, and atom k acts as a nucleophilic species.
Finally, it should be noted that Domingo proposed the Parr functions P(r) [32,33], whose definitions are given by the following equations:
P- (r)= ρsrc (9)
for electrophilic attacks, and
P+ (r)= ρsra (10)
for nucleophilic attacks, which are related to the atomic spin density (ASD) at the r atom of the radical cation or anion of a given molecule, respectively [34].
Following the lines of our previous works [2–9], the computational studies were performed with the Gaussian 09 [35] series of programs with density functional methods as implemented in the computational package. The basis sets used in this work were Def2SVP for geometry optimization and frequencies, whereas Def2TZVP was considered for the calculation of the electronic properties [36,37]. All the calculations were performed in the presence of water as a solvent by performing integral equation formalism-polarized continuum model (IEF-PCM) computations according to the solvation model density (SMD) solvation model [38].
For the calculation of the molecular structures and properties of the studied systems, we chose ten density functionals that are known to consistently provide satisfactory results for several structural and thermodynamic properties:
|
CAM-B3LYP |
Long-range-corrected B3LYP by the CAM method |
[39] |
|
LC-ωPBE |
Long-range-corrected ωPBE density functional |
[40] |
|
M11 |
Range-separated hybrid meta-GGA |
[41] |
|
M11L |
Dual-range local meta-GGA |
[42] |
|
MN12L |
Non-separable local meta-NGA |
[43] |
|
MN12SX |
Range-separated hybrid non-separable meta-NGA |
[44] |
|
N12 |
Non-separable local NGA |
[45] |
|
N12SX |
Range-separated hybrid NGA |
[44] |
|
ωB97X |
Long-range corrected density functional |
[46] |
|
ωB97XD |
ωB97X version including empirical dispersion |
[47] |
In these functionals, GGA stands for generalized gradient approximation (in which the density functional depends on the up and down spin densities and their reduced gradient) and NGA stands for non-separable gradient approximation (in which the density functional depends on the up and down spin densities and their reduced gradient, and also adopts a non-separable form).
This study took the molecular structures of the Red-M1 intermediate melanoidin pigment from PubChem (https://pubchem.ncbi.nlm.nih.gov), a public repository for information pertaining chemical substances together with the biological activities with which they are associated. The molecular structure with IUPAC Name is 1,4,6,9-tetracarboxymethyl-5-(1,2,3,4-tetrahydroxybutyl)-8-hydroxymethyl-3-(2,3dihydroxy-propyl)-5,6-dihydro-pyrrolo[2’,3’:4,5]pyrrolo[2,3-e]pyrrolo[3,2-b]azepine-9-ium.Red-M2 was built as the diastereomer of the Red-M1 molecule. The pre- optimization of the resultant system involved selecting the most stable conformers. The selection was done using random sampling that involved molecular mechanics techniques and the inclusion of various torsional angles via the general MMFF94 force field [48–52] using the Marvin View 17.15 program, which is an advanced chemical viewer suited to multiple- and single-chemical queries, structures, and re- actions (https://www.chemaxon.com). Afterwards, the structures that the resultant lower-energy conformer assumed for each molecule were reoptimized using the ten density functionals mentioned in the previous section, together with the Def2SVP basis set as well as the SMD solvation model, in which the solvent was water.
The analysis of the results obtained in the study aimed at verifying that the KID procedure was fulfilled. On doing it previously, several descriptors associated with the results that the HOMO and LUMO calculations obtained are related with results obtained using the vertical I and A following the ∆SCF procedure. A link exists between the three main descriptors and the simplest conformity to the Koopmans’ theorem by linking εH with -I, εL with -A, and their behavior in describing the HOMO–LUMO gap as JI=|εH+Egs (N-1)-Egs (N)|, JA=|εL+Egs (N)-Egs (N+1)| , and JHL=√((JI2+JA2 ) ). Notably, the JA descriptor consists of an approximation that remains valid only when the HOMO that a radical anion has (the SOMO) shares similarity with the LUMO of the neutral system. Consequently, we decided to design another descriptor ∆SL, to guide in verifying the accuracy of the approximation.
Tables 1 and 2 illustrate the electronic energies of neutral, positive, and negative molecular systems of the Red-M1 and Red-M2 intermediate melanoidins; the HOMO, LUMO, and SOMO orbital energies (all in au); and the calculation of the JI, JA, JHL, and ∆SL descriptors involves using the ten density functional as well as the Def2TZVP basis set with water as a solvent, which is simulated through the SMD parametrization of the IEF-PCM model.
Table 1. Electronic energies of the neutral, positive and negative molecular systems (in au) of Red-M1; the HOMO, LUMO and SOMO orbital energies (also in au); the JI , JA , JHL and ∆SL descriptors calculated with the ten density functionals and the Def2TZVP basis set using water as a solvent, simulated with the SMD parametrization of the IEF-PCM model.
|
|
E0 |
E+ |
E- |
HOMO |
LUMO |
SOMO |
JI |
JA |
JHL |
∆SL |
|
CAM-B3LYP |
-2432.5212 |
-2432.3343 |
-2432.6334 |
-0.2360 |
-0.0626 |
-0.1607 |
0.0490 |
0.0496 |
0.0697 |
0.0982 |
|
LC-ωPBE |
-2432.0877 |
-2431.8951 |
-2432.2131 |
-0.2797 |
-0.0389 |
-0.2092 |
0.0871 |
0.0865 |
0.1227 |
0.1703 |
|
M11 |
-2432.3786 |
-2432.1828 |
-2432.4990 |
-0.2739 |
-0.0431 |
-0.1961 |
0.0781 |
0.0774 |
0.1100 |
0.1531 |
|
M11L |
-2432.2581 |
-2432.0594 |
-2432.3776 |
-0.1905 |
-0.1296 |
-0.1101 |
0.0082 |
0.0101 |
0.0130 |
0.0196 |
|
MN12L |
-2431.3635 |
-2431.1766 |
-2431.4687 |
-0.1792 |
-0.1136 |
-0.0979 |
0.0077 |
0.0085 |
0.0115 |
0.0157 |
|
MN12SX |
-2431.5013 |
-2431.3063 |
-2431.6142 |
-0.1944 |
-0.1130 |
-0.1126 |
0.0006 |
0.0001 |
0.0006 |
0.0004 |
|
N12 |
-2433.1637 |
-2432.9889 |
-2433.2615 |
-0.1654 |
-0.1093 |
-0.0880 |
0.0095 |
0.0115 |
0.0149 |
0.0214 |
|
N12SX |
-2432.4786 |
-2432.2928 |
-2432.5870 |
-0.1866 |
-0.1068 |
-0.1094 |
0.0008 |
0.0015 |
0.0017 |
0.0026 |
|
ωB97X |
-2433.0096 |
-2432.8204 |
-2433.1264 |
-0.2692 |
-0.0370 |
-0.1949 |
0.0800 |
0.0798 |
0.1129 |
0.1579 |
|
ωB97XD |
-2432.8597 |
-2432.6701 |
-2432.9735 |
-0.2583 |
-0.0444 |
-0.1822 |
0.0687 |
0.0694 |
0.0977 |
0.1378 |
Table 2. Electronic energies of the neutral, positive and negative molecular systems (in au) of Red-M2; the HOMO, LUMO and SOMO orbital energies (also in au); the JI , JA , JHL and ∆SL descriptors calculated with the ten density functionals and the Def2TZVP basis set using water as a solvent, simulated with the SMD parametrization of the IEF-PCM model.
|
|
E0 |
E+ |
E- |
HOMO |
LUMO |
SOMO |
JI |
JA |
JHL |
∆SL |
|
CAM-B3LYP |
-2432.5212 |
-2432.3343 |
-2432.6334 |
-0.2360 |
-0.0626 |
-0.1607 |
0.0490 |
0.0496 |
0.0697 |
0.0982 |
|
LC-ωPBE |
-2432.0877 |
-2431.8951 |
-2432.2131 |
-0.2797 |
-0.0389 |
-0.2092 |
0.0871 |
0.0865 |
0.1227 |
0.1703 |
|
M11 |
-2432.3786 |
-2432.1828 |
-2432.4990 |
-0.2739 |
-0.0431 |
-0.1961 |
0.0781 |
0.0774 |
0.1100 |
0.1531 |
|
M11L |
-2432.2581 |
-2432.0594 |
-2432.3776 |
-0.1905 |
-0.1296 |
-0.1101 |
0.0082 |
0.0101 |
0.0130 |
0.0196 |
|
MN12L |
-2431.3636 |
-2431.1766 |
-2431.4687 |
-0.1792 |
-0.1136 |
-0.0979 |
0.0077 |
0.0085 |
0.0115 |
0.0157 |
|
MN12SX |
-2431.5013 |
-2431.3063 |
-2431.6142 |
-0.1944 |
-0.1130 |
-0.1126 |
0.0006 |
0.0001 |
0.0006 |
0.0004 |
|
N12 |
-2433.1637 |
-2432.9889 |
-2433.2615 |
-0.1654 |
-0.1093 |
-0.0880 |
0.0095 |
0.0115 |
0.0149 |
0.0213 |
|
N12SX |
-2432.4786 |
-2432.2928 |
-2432.5870 |
-0.1866 |
-0.1068 |
-0.1094 |
0.0008 |
0.0015 |
0.0017 |
0.0026 |
|
ωB97X |
-2433.0096 |
-2432.8204 |
-2433.1263 |
-0.2692 |
-0.0370 |
-0.1949 |
0.0800 |
0.0798 |
0.1129 |
0.1579 |
|
ωB97XD |
-2432.8597 |
-2432.6701 |
-2432.9735 |
-0.2583 |
-0.0444 |
-0.1822 |
0.0687 |
0.0694 |
0.0976 |
0.1378 |
Next, we consider four other descriptors that analyze how useful the studied density functionals are for the prediction of the electronegativity χ, global hardness η, and global electrophilicity ω. For a combination of these conceptual DFT descriptors, considering only the energies of the HOMO and LUMO or the vertical I and A:Jχ=|χ-χK |, Jη=|η-ηK |, and JCDFT=√((Jχ2+Jη2+Jω2 ) ), where CDFT stands for conceptual DFT. The results of the calculations of Jχ, Jη, Jω, and JCDFT for the low-energy conformers of the Red-M1 and Red-M2 intermediate melanoidin pigments are displayed in Tables 3 and 4.
Table 3. Jχ, Jη, Jω, and JCDFT for the Red-M1 intermediate melanoidin pigment.
|
|
Jχ |
Jη |
Jω |
JCDFT |
|
CAM-B3LYP |
0.0003 |
0.0986 |
0.0852 |
0.1303 |
|
LC-ωPBE |
0.0003 |
0.1736 |
0.1354 |
0.2201 |
|
M11 |
0.0004 |
0.1555 |
0.1115 |
0.1914 |
|
M11L |
0.0010 |
0.0183 |
0.0505 |
0.0538 |
|
MN12L |
0.0004 |
0.0162 |
0.0329 |
0.0367 |
|
MN12SX |
0.0003 |
0.0007 |
0.0008 |
0.0011 |
|
N12 |
0.0010 |
0.0210 |
0.0477 |
0.0521 |
|
N12SX |
0.0004 |
0.0024 |
0.0048 |
0.0054 |
|
ωB97X |
0.0001 |
0.1597 |
0.1109 |
0.1945 |
|
ωB97XD |
0.0004 |
0.1381 |
0.0983 |
0.1695 |
Table 4. Jχ, Jη, Jω, and JCDFT for the Red-M2 intermediate melanoidin pigment.
|
|
Jχ |
Jη |
Jω |
JCDFT |
|
CAM-B3LYP |
0.0003 |
0.0986 |
0.0852 |
0.1303 |
|
LC-ωPBE |
0.0003 |
0.1736 |
0.1354 |
0.2201 |
|
M11 |
0.0004 |
0.1555 |
0.1115 |
0.1914 |
|
M11L |
0.0010 |
0.0183 |
0.0506 |
0.0538 |
|
MN12L |
0.0004 |
0.0162 |
0.0329 |
0.0367 |
|
MN12SX |
0.0003 |
0.0007 |
0.0008 |
0.0011 |
|
N12 |
0.0010 |
0.0210 |
0.0476 |
0.0521 |
|
N12SX |
0.0004 |
0.0023 |
0.0048 |
0.0054 |
|
ωB97X |
0.0001 |
0.1597 |
0.1109 |
0.1945 |
|
ωB97XD |
0.0004 |
0.1381 |
0.0983 |
0.1695 |
As Tables 1 to 4 show, the KID procedure applies accurately from the MN12SX and N12SX density functionals that are RS hybrid meta-NGA and RS hybrid NGA density functionals, respectively. In fact, the values of JI, JA, and JHL are not zero. Nevertheless, the results tend to be impressive, especially for the MN12SX density functional. Additionally, the ∆SL descriptor reaches the minimum values when the MN12SX and N12SX density functionals are used in the calculations. This implies that there is sufficient justification to assume that the LUMO of the neutral approximates the electron affinity.
The same density functionals follow the KID procedure in the rest of the descriptors, such as Jχ, Jη, Jω, and . The significance of these results is attributable to their illustration that reliance on JI, JA, and JHL would not be sufficient. For instance, if Jχ were considered on its own to apply to each density functional covered in this study, the values would be considerably near zero. In the case of the other descriptors, only the MN12SX and N12SX density functionals exhibit this behavior. This implies that the results from Jχ are likely to be because of the cancellation of errors done by chance.
The Red-M1 and Red-M2 intermediate melanoidin pigments would be best studied using the time-dependent DFT (TDDFT), because the pigments are colored molecules. In the past, various TDDFT studies of molecules of different size have used optimally tuned RSH density functionals [23,53–71].
The considerable success of this approach is however undermined by the issue of tuning being system-dependent. Therefore, the focus should be on establishing the effectiveness of the behaviors of the fixed RSH density functionals in describing the excitation characteristics. Becke has recently mentioned that the adiabatic connection and the ideas of Hohenberg, Kohn, and Sham applying only to electronic ground states is a common misconception [72]. Furthermore, consistent with Baerends et al., the KS model is not appreciated for being superior because of its lowest excitation energy in molecules. Physically, this amounts to an excitation of the KS system rather than electron addition, as would be the case in Hartree–Fock. Thus, it can be effectively used as a measure of the optical gap and is an effective approximation to the gap (in molecules) [73]. In their conclusion, van Meer et al. proposed that the HOMO–LUMO gap associated with the KS model tends to be an approximation of the lowest excitation energy, which is a desirable characteristic with no concerns regarding it [74].
Therefore, the calculation of the maximum wavelength absorption of the Red-M1 and Red-M2 pigments involved conducting ground-state calculations with the aforementioned ten density functionals at the same level of model chemistry and theory and determining the HOMO–LUMO gap. Figure 1 provides an illustration that graphically compares the results involved in the ground-state approximation derived from the HOMO–LUMO gap together with the experimental value of 564 nm for the Red-M1 molecule and 554 nm for the Red-M2 isomer [15].
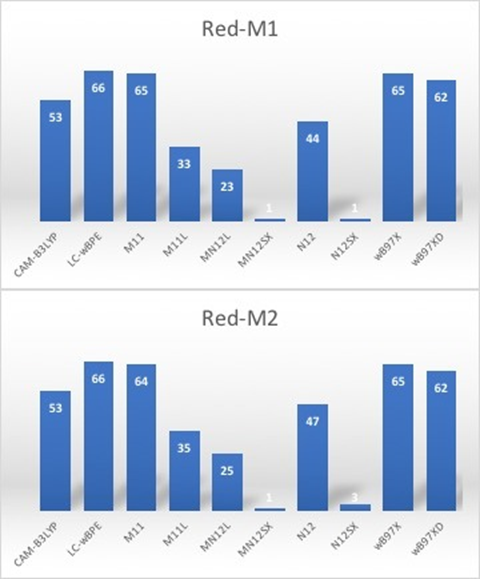
Figure 1. Graphical comparison of the results for the calculation of the λmax of the Red-M1 and Red-M2 pigments between the HOMO-LUMO gap and the experimental data.
Notably, the presented results suggest that the differences with the experimental value for λmax tend to have the same order in the various functionals that the current study considers, apart from the N12 density functional. If the λmax values that the HOMO–LUMO gap generates were those considered, MN12SX and N12SX would appear to be especially accurate in predicting this value. This finding does not apply to the rest of the density functionals that this study considered.
These results can be explained by considering that both functionals have been built as the screened exchange (SX) version of range separation in which the electron exchange for small interelectronic distances is treated with a finite percentage of nonlocal HF exchange, but the nonlocality is screened at large distances, and electron exchange at long range is treated by a local approximation. This can be justified on the physical grounds that nonlocal exchange may be screened at long range by correlation effects and make those functionals as the perfect choice for predicting outer valence and Rydberg excitations. Moreover, it can be seen from Tables 1 to 4 that the values of the descriptors are very small in these cases and also the ∆SL values are also small meaning that the values of the energy of the LUMO are predicted accurately in addition to the HOMO energies.
Upon verifying that the MN12SX density functional has the tendency of peaking in calculating the global reactivity density descriptors and in predicting λmax, Figures 2 and 3 graphically present the optimized structures of the Red-M1 and Red-M2 pigments, as calculated based on the theory. Tables 5 and 7 illustrate the bond lengths, and Tables 6 and 8 demonstrate the bond angles.
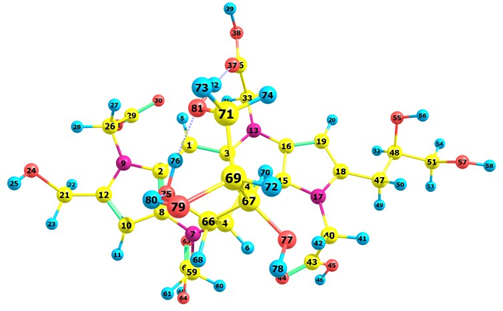
Figure 2. Schematic representation of the optimized structure of the Red-M1 pigment calculated with the MN12SX density functional showing the numbering of the atoms.
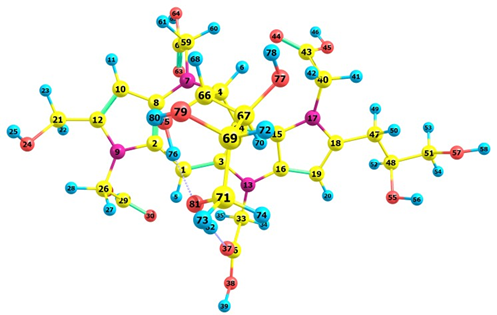
Figure 3. Schematic representation of the optimized structure of the Red-M2 pigment calculated with the MN12SX density functional showing the numbering of the atoms.
Table 5. Calculated bond lengths (in A) of the Red-M1 intermediate melanoidin pigment with the MN12SX density functional.
|
Bond |
Distance |
Bond |
Distance |
Bond |
Distance |
Bond |
Distance |
|
R (1-2) |
1.383 |
R (15-16) |
1.417 |
R (36-38) |
1.361 |
R (62-63) |
1.219 |
|
R (1-3) |
1.406 |
R (15-17) |
1.386 |
R (38-39) |
0.982 |
R (62-64) |
1.372 |
|
R (1-5) |
1.103 |
R (16-19) |
1.414 |
R (40-41) |
1.107 |
R (64-65) |
0.982 |
|
R (2-8) |
1.450 |
R (17-18) |
1.384 |
R (40-42) |
1.117 |
R (66-67) |
1.540 |
|
R (2-9) |
1.402 |
R (17-40) |
1.447 |
R (40-43) |
1.524 |
R (66-68) |
1.124 |
|
R (3-13) |
1.400 |
R (18-19) |
1.397 |
R (43-44) |
1.226 |
R (66-75) |
1.442 |
|
R (3-14) |
1.429 |
R (18-47) |
1.497 |
R (43-45) |
1.365 |
R (67-69) |
1.545 |
|
R (4-6) |
1.118 |
R (19-20) |
1.093 |
R (45-46) |
0.983 |
R (67-70) |
1.122 |
|
R (4-7) |
1.464 |
R (21-22) |
1.114 |
R (47-48) |
1.536 |
R (67-77) |
1.457 |
|
R (4-14) |
1.513 |
R (21-23) |
1.114 |
R (47-49) |
1.112 |
R (69-71) |
1.536 |
|
R (4-66) |
1.559 |
R (21-24) |
1.438 |
R (47-50) |
1.112 |
R (69-72) |
1.120 |
|
R (7-8) |
1.362 |
R (24-25) |
0.981 |
R (48-51) |
1.525 |
R (69-79) |
1.442 |
|
R (7-59) |
1.451 |
R (26-27) |
1.111 |
R (48-52) |
1.120 |
R (71-73) |
1.115 |
|
R (8-10) |
1.415 |
R (26-28) |
1.109 |
R (48-55) |
1.443 |
R (71-74) |
1.114 |
|
R (9-12) |
1.362 |
R (26-29) |
1.527 |
R (51-53) |
1.121 |
R (71-81) |
1.447 |
|
R (9-26) |
1.449 |
R (29-30) |
1.225 |
R (51-54) |
1.117 |
R (75-76) |
1.003 |
|
R (10-11) |
1.093 |
R (29-31) |
1.363 |
R (51-57) |
1.438 |
R (77-78) |
0.983 |
|
R (10-12) |
1.395 |
R (31-32) |
0.983 |
R (55-56) |
0.985 |
R (79-80) |
0.988 |
|
R (12-21) |
1.504 |
R (33-34) |
1.108 |
R (57-58) |
0.981 |
R (81-82) |
0.987 |
|
R (13-16) |
1.369 |
R (33-35) |
1.113 |
R (59-60) |
1.109 |
R (37-82) |
1.940 |
|
R (13-33) |
1.441 |
R (33-36) |
1.522 |
R (59-61) |
1.118 |
R (75-80) |
1.965 |
|
R (14-15) |
1.403 |
R (36-37) |
1.227 |
R (59-62) |
1.522 |
R (76-81) |
1.700 |
Table 6. Calculated bond angles (in o) of the Red-M1 intermediate melanoidin pigment with the MN12SX density functional.
|
Bond |
Angle |
Bond |
Angle |
Bond |
Angle |
Bond |
Angle |
|
A (2-1-3) |
123.6 |
A (9-26-28) |
108.0 |
A (26-29-31) |
114.4 |
A (63-62-64) |
121.6 |
|
A (2-1-5) |
118.0 |
A (9-26-29) |
110.4 |
A (30-29-31) |
121.6 |
A (62-64-65) |
108.4 |
|
A (1-2-8) |
130.7 |
A (11-10-12) |
124.9 |
A (29-31-32) |
108.8 |
A (67-66-68) |
109.3 |
|
A (1-2-9) |
122.3 |
A (10-12-21) |
128.0 |
A (34-33-35) |
107.3 |
A (67-66-75) |
112.0 |
|
A (3-1-5) |
118.1 |
A (12-21-22) |
110.0 |
A (34-33-36) |
109.7 |
A (66-67-79) |
115.2 |
|
A (1-3-13) |
122.3 |
A (12-21-23) |
109.6 |
A (35-33-36) |
109.2 |
A (66-67-70) |
109.2 |
|
A (1-3-14) |
128.7 |
A (12-21-24) |
111.0 |
A (33-36-37) |
124.8 |
A (66-67-77) |
107.4 |
|
A (8-2-9) |
105.8 |
A (16-13-33) |
125.7 |
A (33-36-38) |
113.8 |
A (68-66-75) |
107.2 |
|
A (2-8-7) |
127.4 |
A (13-16-15) |
108.1 |
A (37-36-38) |
121.4 |
A (66-75-76) |
108.0 |
|
A (2-8-10) |
106.9 |
A (13-16-19) |
143.2 |
A (36-37-82) |
163.0 |
A (66-75-80) |
90.1 |
|
A (2-9-12) |
110.3 |
A (13-33-34) |
108.8 |
A (36-38-39) |
109.0 |
A (69-67-70) |
108.9 |
|
A (2-9-26) |
124.1 |
A (13-33-35) |
110.6 |
A (41-40-42) |
108.8 |
A (69-67-77) |
109.7 |
|
A (13-3-14) |
108.9 |
A (13-33-36) |
111.1 |
A (41-40-43) |
110.0 |
A (67-69-71) |
112.1 |
|
A (3-13-16) |
108.5 |
A (14-15-16) |
109.1 |
A (42-40-43) |
109.9 |
A (67-69-72) |
108.2 |
|
A (3-13-33) |
125.8 |
A (14-15-17) |
143.6 |
A (40-43-44) |
124.6 |
A (67-69-79) |
111.1 |
|
A (3-14-4) |
127.2 |
A (16-15-17) |
107.2 |
A (40-43-45) |
114.3 |
A (70-67-77) |
106.1 |
|
A (3-14-15) |
105.4 |
A (15-16-19) |
108.7 |
A (44-43-45) |
121.1 |
A (67-77-78) |
107.4 |
|
A (6-4-7) |
106.8 |
A (15-17-18) |
108.2 |
A (43-45-46) |
108.7 |
A (71-69-72) |
107.6 |
|
A (6-4-14) |
106.9 |
A (15-17-40) |
125.0 |
A (48-47-49) |
109.4 |
A (71-69-79) |
111.2 |
|
A (6-4-66) |
107.6 |
A (16-19-18) |
105.6 |
A (48-47-50) |
108.9 |
A (69-71-73) |
110.0 |
|
A (7-4-14) |
111.3 |
A (16-19-20) |
128.8 |
A (47-48-51) |
111.0 |
A (69-71-74) |
109.6 |
|
A (7-4-66) |
110.2 |
A (18-17-40) |
126.7 |
A (47-48-52) |
109.5 |
A (69-71-81) |
110.7 |
|
A (4-7-8) |
122.8 |
A (17-18-19) |
110.2 |
A (47-48-55) |
109.9 |
A (72-69-79) |
106.4 |
|
A (4-7-59) |
117.6 |
A (17-18-47) |
121.2 |
A (49-47-50) |
106.2 |
A (69-79-80) |
107.3 |
|
A (14-4-66) |
113.6 |
A (17-40-41) |
108.7 |
A (51-48-52) |
110.1 |
A (73-71-74) |
108.0 |
|
A (4-14-15) |
127.2 |
A (17-40-42) |
109.1 |
A (51-48-55) |
108.4 |
A (73-71-81) |
109.5 |
|
A (4-66-67) |
110.7 |
A (17-40-43) |
110.2 |
A (48-51-53) |
110.9 |
A (74-71-81) |
108.9 |
|
A (4-66-68) |
107.5 |
A (19-18-47) |
128.5 |
A (48-51-54) |
110.4 |
A (71-81-82) |
109.9 |
|
A (4-66-75) |
110.1 |
A (18-19-20) |
125.6 |
A (48-51-57) |
107.3 |
A (71-81-76) |
111.2 |
|
A (8-7-59) |
118.2 |
A (18-47-48) |
112.8 |
A (52-48-55) |
107.8 |
A (76-75-80) |
82.5 |
|
A (7-8-10) |
125.6 |
A (18-47-49) |
109.5 |
A (48-55-56) |
105.9 |
A (75-76-81) |
160.9 |
|
A (7-59-60) |
109.2 |
A (18-47-50) |
109.7 |
A (53-51-54) |
107.6 |
A (79-80-75) |
134.0 |
|
A (7-59-61) |
111.4 |
A (22-21-23) |
108.1 |
A (53-51-57) |
110.4 |
A (81-82-37) |
155.9 |
|
A (7-59-62) |
110.5 |
A (22-21-24) |
107.5 |
A (54-51-57) |
110.0 |
A (82-81-76) |
126.4 |
|
A (8-10-11) |
127.1 |
A (23-21-24) |
110.6 |
A (51-57-58) |
108.1 |
|
|
|
A (8-10-12) |
107.9 |
A (21-24-25) |
108.4 |
A (60-59-61) |
107.1 |
|
|
|
A (12-9-26) |
125.7 |
A (27-26-28) |
108.8 |
A (60-59-62) |
108.9 |
|
|
|
A (9-12-10) |
109.0 |
A (27-26-29) |
109.4 |
A (61-59-62) |
109.6 |
|
|
|
A (9-12-21) |
123.0 |
A (28-26-29) |
111.1 |
A (59-62-63) |
125.0 |
|
|
|
A (9-26-27) |
109.1 |
A (26-29-30) |
124.0 |
A (59-62-64) |
113.4 |
|
|
Table 7. Calculated bond lengths (in A) of the Red-M2 intermediate melanoidin pigment with the MN12SX density functional.
|
Bond |
Distance |
Bond |
Distance |
Bond |
Distance |
Bond |
Distance |
|
R (1-2) |
1.383 |
R (15-16) |
1.417 |
R (36-38) |
1.361 |
R (62-63) |
1.219 |
|
R (1-3) |
1.406 |
R (15-17) |
1.386 |
R (38-39) |
0.982 |
R (62-64) |
1.372 |
|
R (1-5) |
1.103 |
R (16-19) |
1.414 |
R (40-41) |
1.107 |
R (64-65) |
0.982 |
|
R (2-8) |
1.450 |
R (17-18) |
1.384 |
R (40-42) |
1.117 |
R (66-67) |
1.540 |
|
R (2-9) |
1.402 |
R (17-40) |
1.447 |
R (40-43) |
1.524 |
R (66-68) |
1.124 |
|
R (3-13) |
1.400 |
R (18-19) |
1.397 |
R (43-44) |
1.226 |
R (66-75) |
1.442 |
|
R (3-14) |
1.429 |
R (18-47) |
1.497 |
R (43-45) |
1.365 |
R (67-69) |
1.545 |
|
R (4-6) |
1.118 |
R (19-20) |
1.093 |
R (45-46) |
0.983 |
R (67-70) |
1.122 |
|
R (4-7) |
1.464 |
R (21-22) |
1.114 |
R (47-48) |
1.536 |
R (67-77) |
1.457 |
|
R (4-14) |
1.513 |
R (21-23) |
1.114 |
R (47-49) |
1.112 |
R (69-71) |
1.536 |
|
R (4-66) |
1.559 |
R (21-24) |
1.438 |
R (47-50) |
1.112 |
R (69-72) |
1.120 |
|
R (7-8) |
1.362 |
R (24-25) |
0.981 |
R (48-51) |
1.525 |
R (69-79) |
1.442 |
|
R (7-59) |
1.451 |
R (26-27) |
1.111 |
R (48-52) |
1.120 |
R (71-73) |
1.115 |
|
R (8-10) |
1.415 |
R (26-28) |
1.109 |
R (48-55) |
1.443 |
R (71-74) |
1.114 |
|
R (9-12) |
1.362 |
R (26-29) |
1.527 |
R (51-53) |
1.121 |
R (71-81) |
1.447 |
|
R (9-26) |
1.449 |
R (29-30) |
1.225 |
R (51-54) |
1.117 |
R (75-76) |
1.003 |
|
R (10-11) |
1.093 |
R (29-31) |
1.363 |
R (51-57) |
1.438 |
R (77-78) |
0.983 |
|
R (10-12) |
1.395 |
R (31-32) |
0.983 |
R (55-56) |
0.985 |
R (79-80) |
0.988 |
|
R (12-21) |
1.504 |
R (33-34) |
1.108 |
R (57-58) |
0.981 |
R (81-82) |
0.987 |
|
R (13-16) |
1.369 |
R (33-35) |
1.113 |
R (59-60) |
1.109 |
R (37-82) |
1.940 |
|
R (13-33) |
1.441 |
R (33-36) |
1.522 |
R (59-61) |
1.118 |
R (75-80) |
1.965 |
|
R (14-15) |
1.403 |
R (36-37) |
1.227 |
R (59-62) |
1.522 |
R (76-81) |
1.700 |
Table 8. Calculated bond angles (in o) of the Red-M2 intermediate melanoidin pigment with the MN12SX density functional.
|
Bond |
Angle |
Bond |
Angle |
Bond |
Angle |
Bond |
Angle |
|
A (2-1-3) |
123.6 |
A (9-26-28) |
108.0 |
A (26-29-31) |
114.4 |
A (63-62-64) |
121.6 |
|
A (2-1-5) |
118.0 |
A (9-26-29) |
110.4 |
A (30-29-31) |
121.6 |
A (62-64-65) |
108.4 |
|
A (1-2-8) |
130.7 |
A (11-10-12) |
124.9 |
A (29-31-32) |
108.8 |
A (67-66-68) |
109.3 |
|
A (1-2-9) |
122.3 |
A (10-12-21) |
128.0 |
A (34-33-35) |
107.3 |
A (67-66-75) |
112.0 |
|
A (3-1-5) |
118.1 |
A (12-21-22) |
110.0 |
A (34-33-36) |
109.7 |
A (66-67-79) |
115.2 |
|
A (1-3-13) |
122.3 |
A (12-21-23) |
109.6 |
A (35-33-36) |
109.2 |
A (66-67-70) |
109.2 |
|
A (1-3-14) |
128.7 |
A (12-21-24) |
111.0 |
A (33-36-37) |
124.8 |
A (66-67-77) |
107.4 |
|
A (8-2-9) |
105.8 |
A (16-13-33) |
125.7 |
A (33-36-38) |
113.8 |
A (68-66-75) |
107.2 |
|
A (2-8-7) |
127.4 |
A (13-16-15) |
108.1 |
A (37-36-38) |
121.4 |
A (66-75-76) |
108.0 |
|
A (2-8-10) |
106.9 |
A (13-16-19) |
143.2 |
A (36-37-82) |
163.0 |
A (66-75-80) |
90.1 |
|
A (2-9-12) |
110.3 |
A (13-33-34) |
108.8 |
A (36-38-39) |
109.0 |
A (69-67-70) |
108.9 |
|
A (2-9-26) |
124.1 |
A (13-33-35) |
110.6 |
A (41-40-42) |
108.8 |
A (69-67-77) |
109.7 |
|
A (13-3-14) |
108.9 |
A (13-33-36) |
111.1 |
A (41-40-43) |
110.0 |
A (67-69-71) |
112.1 |
|
A (3-13-16) |
108.5 |
A (14-15-16) |
109.1 |
A (42-40-43) |
109.9 |
A (67-69-72) |
108.2 |
|
A (3-13-33) |
125.8 |
A (14-15-17) |
143.6 |
A (40-43-44) |
124.6 |
A (67-69-79) |
111.1 |
|
A (3-14-4) |
127.2 |
A (16-15-17) |
107.2 |
A (40-43-45) |
114.3 |
A (70-67-77) |
106.1 |
|
A (3-14-15) |
105.4 |
A (15-16-19) |
108.7 |
A (44-43-45) |
121.1 |
A (67-77-78) |
107.4 |
|
A (6-4-7) |
106.8 |
A (15-17-18) |
108.2 |
A (43-45-46) |
108.7 |
A (71-69-72) |
107.6 |
|
A (6-4-14) |
106.9 |
A (15-17-40) |
125.0 |
A (48-47-49) |
109.4 |
A (71-69-79) |
111.2 |
|
A (6-4-66) |
107.6 |
A (16-19-18) |
105.6 |
A (48-47-50) |
108.9 |
A (69-71-73) |
110.0 |
|
A (7-4-14) |
111.3 |
A (16-19-20) |
128.8 |
A (47-48-51) |
111.0 |
A (69-71-74) |
109.6 |
|
A (7-4-66) |
110.2 |
A (18-17-40) |
126.7 |
A (47-48-52) |
109.5 |
A (69-71-81) |
110.7 |
|
A (4-7-8) |
122.8 |
A (17-18-19) |
110.2 |
A (47-48-55) |
109.9 |
A (72-69-79) |
106.4 |
|
A (4-7-59) |
117.6 |
A (17-18-47) |
121.2 |
A (49-47-50) |
106.2 |
A (69-79-80) |
107.3 |
|
A (14-4-66) |
113.6 |
A (17-40-41) |
108.7 |
A (51-48-52) |
110.1 |
A (73-71-74) |
108.0 |
|
A (4-14-15) |
127.2 |
A (17-40-42) |
109.1 |
A (51-48-55) |
108.4 |
A (73-71-81) |
109.5 |
|
A (4-66-67) |
110.7 |
A (17-40-43) |
110.2 |
A (48-51-53) |
110.9 |
A (74-71-81) |
108.9 |
|
A (4-66-68) |
107.5 |
A (19-18-47) |
128.5 |
A (48-51-54) |
110.4 |
A (71-81-82) |
109.9 |
|
A (4-66-75) |
110.1 |
A (18-19-20) |
125.6 |
A (48-51-57) |
107.3 |
A (71-81-76) |
111.2 |
|
A (8-7-59) |
118.2 |
A (18-47-48) |
112.8 |
A (52-48-55) |
107.8 |
A (76-75-80) |
82.5 |
|
A (7-8-10) |
125.6 |
A (18-47-49) |
109.5 |
A (48-55-56) |
105.9 |
A (75-76-81) |
160.9 |
|
A (7-59-60) |
109.2 |
A (18-47-50) |
109.7 |
A (53-51-54) |
107.6 |
A (79-80-75) |
134.0 |
|
A (7-59-61) |
111.4 |
A (22-21-23) |
108.1 |
A (53-51-57) |
110.4 |
A (81-82-37) |
155.9 |
|
A (7-59-62) |
110.5 |
A (22-21-24) |
107.5 |
A (54-51-57) |
110.0 |
A (82-81-76) |
126.4 |
|
A (8-10-11) |
127.1 |
A (23-21-24) |
110.6 |
A (51-57-58) |
108.1 |
|
|
|
A (8-10-12) |
107.9 |
A (21-24-25) |
108.4 |
A (60-59-61) |
107.1 |
|
|
|
A (12-9-26) |
125.7 |
A (27-26-28) |
108.8 |
A (60-59-62) |
108.9 |
|
|
|
A (9-12-10) |
109.0 |
A (27-26-29) |
109.4 |
A (61-59-62) |
109.6 |
|
|
|
A (9-12-21) |
123.0 |
A (28-26-29) |
111.1 |
A (59-62-63) |
125.0 |
|
|
|
A (9-26-27) |
109.1 |
A (26-29-30) |
124.0 |
A (59-62-64) |
113.4 |
|
|
The electrodonating (ω−) and electroaccepting (ω+) powers have been defined as follows [75]:
ω-=(3I+A)2/(16(I-A))≈(3εH+εL)2/(16ηK ) (11)
and
ω+=(I+3A)2/(16(I-A))≈(εH+3εL)2/(16ηK ) (12)
given that ηK is equal to the right side of Equation 4.
Consequently, a higher ω+ value is associated with improved inclination to accepting change. In contrast, a lower ω− value represents improvement of the system as an electron donor. For comparisons of ω+ and -ω−, the proposed definition of the net electrophilicity includes [76]:
∆ω±= ω+-(-ω- )= ω++ω- (13)
which represents the electroaccepting power in relation to the electrodonating power.
Table 9 illustrates the results obtained after calculating the electronegativity χ, chemical hardness η , global electrophilicity ω , electroaccepting ω+, and electrodonating ω- powers, as well as the net electrophilicity with the MN12SX density functional. The Def2TZVP basis set is used with water acting as a solvent, in line with the SMD solvation model. It can be seen that the global descriptors cannot discriminate between the two isomers.
Table 9. Global reactivity descriptors for the Red-M1 and Red-M2 intermediate melanoidin pigments calculated with the MN12SX density functional.
|
|
Electronegativity (χ) |
Chemical Hardness (η) |
Electrophilicity (ω) |
|
Red-M1 |
4.1816 |
2.2154 |
3.9464 |
|
Red-M2 |
4.1816 |
2.2154 |
3.9464 |
|
|
Electrodonating power (ω-) |
Electroaccepting power (ω+) |
Net electrophilicity (∆ω±) |
|
Red-M1 |
5.9520 |
4.5597 |
10.5117 |
|
Red-M2 |
5.9520 |
4.5597 |
10.5117 |
The calculations of the condensed Fukui functions and DD have been done by using the Chemcraft molecular analysis program to extract the Mulliken and NPA atomic charges [77], beginning with single-point energy calculations involving the MN12SX density functional that uses the Def2TZVP basis set, also in line with the SMD solvation model, water is utilized as a solvent.
Considering the potential application of the Red-M1 and Red-M2 molecules as an antioxidant, it is of interest to get insight into the active sites for radical attack. A graphical representation of the radical Fukui function is presented in Figures 4 and 5 for both isomers.
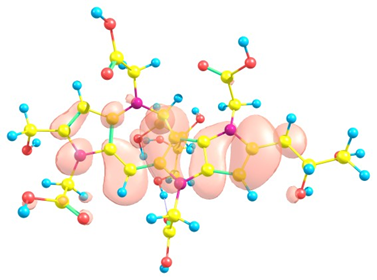
Figure 4. Graphical schematic representation of the radical Fukui function of the Red-M1 intermediate melanoidin pigment.
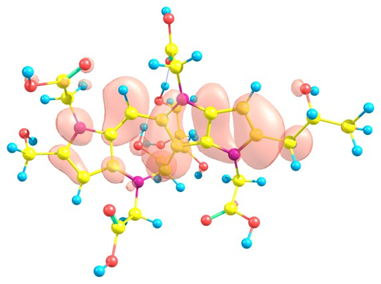
Figure 5. Graphical schematic representation of the radical Fukui function of the Red-M2 intermediate melanoidin pigment.
The condensed electrophilic and nucleophilic Parr functions Pk- and Pk+ over the atoms of the Red-M1 and Red-M2 pigments have been calculated by extracting the Mulliken and Hirshfeld (or CM5) atomic charges using the Chemcraft molecular analysis program [77], starting from single-point energy calculations of the ionic species with the MN12SX density functional using the Def2TZVP basis set in the presence of water as a solvent, according to the SMD solvation model.
The results for the condensed DD calculated with Mulliken and NPA atomic charges, ∆fk (M) and ∆fk (N), as well as the electrophilic and nucleophilic Parr functions with Mulliken atomic charges, Pk-(M), Pk+(M), and with Hirshfeld (or CM5) atomic charges Pk-(H) and Pk+(H) , are displayed in Tables 10 and 11 for Red-M1 and Red-M2, respectively.
Table 10. Condensed dual descriptor calculated with Mulliken atomic charges ∆fk (M) and with NPA atomic charges ∆fk (N), the electrophilic and nucleophilic Parr functions with Mulliken atomic charges, Pk-(M), Pk+(M), and with Hirshfeld (or CM5) atomic charges, Pk-(H) and Pk+(H) for the Red-M1 molecule. Only the values of ∆fk for which the absolute values is greater than 5 are displayed because lower values are not of significance for the chemical reactivity.
|
Atom |
∆fk (M) |
∆fk (N) |
Pk+(M) |
Pk-(M) |
Pk+(H) |
Pk-(H) |
|
1C |
22.24 |
17.19 |
0.4098 |
-0.1634 |
0.2159 |
-0.0401 |
|
2C |
-10.74 |
-10.22 |
-0.0904 |
0.2599 |
0.0184 |
0.1499 |
|
3C |
-13.51 |
-9.69 |
-0.1356 |
0.2527 |
-0.0046 |
0.1421 |
|
8C |
7.07 |
6.76 |
0.2303 |
0.0257 |
0.1276 |
0.0560 |
|
12C |
7.44 |
5.34 |
0.2223 |
0.0625 |
0.1345 |
0.0440 |
|
14C |
6.38 |
5.66 |
0.2390 |
-0.0006 |
0.1120 |
0.0331 |
|
15C |
-5.26 |
-4.85 |
-0.0482 |
0.1110 |
0.0090 |
0.0790 |
|
19C |
-5.75 |
-4.43 |
-0.0495 |
0.0361 |
-0.0039 |
0.0528 |
,
Table 11. Condensed dual descriptor calculated with Mulliken atomic charges ∆fk (M) and with NPA atomic charges ∆fk (N), the electrophilic and nucleophilic Parr functions with Mulliken atomic charges, Pk-(M) and Pk+(M) and with Hirshfeld (or CM5) atomic charges, Pk-(H) and Pk+(H), for the Red-M2 molecule. Only the values of ∆fk for which the absolute values is greater than 1 are displayed because lower values are not of significance for the chemical reactivity.
|
Atom |
∆fk (M) |
∆fk (N) |
Pk+(M) |
Pk-(M) |
Pk+(H) |
Pk-(H) |
|
1C |
22.24 |
17.19 |
0.4098 |
-0.1634 |
0.2159 |
-0.0401 |
|
2C |
-10.74 |
-10.23 |
-0.0904 |
0.2599 |
0.0184 |
0.1499 |
|
3C |
-13.51 |
-9.69 |
-0.1356 |
0.2527 |
-0.0046 |
0.1421 |
|
8C |
7.07 |
6.76 |
0.2303 |
0.0257 |
0.1276 |
0.0560 |
|
12C |
7.43 |
5.34 |
0.2223 |
0.0625 |
0.1345 |
0.0440 |
|
14C |
6.38 |
5.66 |
0.2390 |
-0.0006 |
0.1120 |
0.0330 |
|
15C |
-5.26 |
-4.85 |
-0.0482 |
0.1110 |
0.0090 |
0.0790 |
|
19C |
-5.75 |
-4.44 |
-0.0495 |
0.0361 |
-0.0039 |
0.0528 |
From the results for the local descriptors in Table 10 and 11, it can be concluded that C1 will be the preferred site for a nucleophilic attack and that this atom will act as an electrophilic species in a chemical reaction. In turn, it can be determined that C2 and C3 will be prone to electrophilic attacks and that these atomic sites will act as nucleophilic species in chemical reactions in which the Red-M1 and Red-M2 molecules are involved.
All calculations have been performed in the gas phase also in order to check the effect of the solvent in the prediction of the λmax and the local chemical reactivity of the melanoidins. The main conclusion is that there are not great differences between the results in either case. However, we believe that it is preferably to keep the results in the solvent phase because they are a better representation of the real chemical phenomena.
Unfortunately, the only experimental results available are those for the λmax of the melanoidins Red-M1 and Red-M2. Our predictions for the chemical reactivity should be validated in the future but we believe that the comparisons between the CDFT descriptors (the Fukui functions and the Dual Descriptor) and the MEDT descriptors (the Parr functions) and the good agreement between them is a good reason to make us confidence that our results are very accurate.
The ten fixed RSH density functionals, including CAM-B3LYP, LC-ωPBE, M11, M11L, MN12L, MN12SX, N12, N12SX, ωB97X, and ωB97XD were examined to establish whether they fulfill the empirical KID procedure. The assessment was performed by comparing the values from HOMO and LUMO calculations to those that the ∆SCF technique generates for the Red-M1 and Red-M2 molecules. These are intermediate melanoidin pigments that are of both academic and industrial interest. The study observed that the RS and hybrid meta-NGA MN12SX and N12SX density functionals tend to be the most suited in meeting this goal. In this case, they emerge as suitable alternatives to the density functionals once it is established that the behaviors of the functionals are tuned using a gap-fitting procedure. They also exhibit desirable prospects of how they will benefit future studies in understanding the chemical reactivity of colored melanoidins with larger molecular weights when reducing sugars react with proteins and peptides.
From the results of this work, it becomes evident that it is easy to predict the sites of interaction of the Red-M1 and Red-M2 pigments under study. This would involve having DFT-based reactivity descriptors including Parr functions and DD calculations. Evidently, the descriptors were useful in characterizing and describing the preferred reactive sites. They were also useful in comprehensively explaining the reactivity of the molecules.
Furthermore, it is also possible to predict the maximum absorption wavelength for the Red-M1 and Red-M2 pigments with considerable accuracy. The prediction would involve the MN12SX density functional beginning with the HOMO–LUMO gap instead of TDDFT calculations. This finding is particularly crucial considering its likelihood of being used to inform an alternative determination method for the color of larger systems such as prosthetic chromophore groups. This would become necessary in circumstances in which the TDDFT calculations would be prohibitive.
This work has been partially supported by CIMAV, SC and Consejo Nacional de Ciencia y Tecnología (CONACYT, Mexico) through Grant 219566-2014 for Basic Science Research. Daniel Glossman-Mitnik conducted this work while a Visiting Lecturer at the University of the Balearic Islands from which support is gratefully acknowledged. This work was cofunded by the Ministerio de Economía y Competitividad (MINECO) and the European Fund for Regional Development (FEDER) (CTQ2014-55835-R).
Author’s contributions
Daniel Glossman-Mitnik conceived and designed the research and headed, wrote, and revised the manuscript; J. Frau contributed to the writing and the revision of the article.
Peverati, R., Truhlar, D.G.: Exchange-Correlation Functional with Good Accuracy for Both Structural and Energetic Properties while Depending Only on the Density and Its Gradient. Journal of Chemical Theory and Computation 8(7), 2310–2319 (2012) PMid:26588964
View Article PubMed/NCBIPeverati, R., Truhlar, D.G.: Screened-Exchange Density Functionals with Broad Accuracy for Chemistry and Solid-State Physics. Physical Chemistry Chemical Physics 14(47), 16187–16191 (2012) PMid:23132141
View Article PubMed/NCBIPeverati, R., Truhlar, D.G.: An Improved and Broadly Accurate Local Approximation to the Exchange-Correlation Density Functional: the MN12-L Functional for Electronic Structure Calculations in Chemistry and Physics. Physical Chemistry Chemical Physics 14(38), 13171–13174 (2012) PMid:22910998
View Article PubMed/NCBIPeverati, R., Truhlar, D.G.: M11-L: A Local Density Functional That Provides Improved Accuracy for Electronic Structure Calculations in Chemistry and Physics. The Journal of Physical Chemistry Letters 3(1), 117–124 (2012)
View ArticlePeverati, R., Truhlar, D.G.: Improving the Accuracy of Hybrid Meta-GGA Density Functionals by Range Separation. The Journal of Physical Chemistry Letters 2(21), 2810–2817 (2011)
View ArticleHenderson, T.M., Izmaylov, A.F., Scalmani, G., Scuseria, G.E.: Can Short-Range Hybrids Describe Long-Range-Dependent Properties? The Journal of Chemical Physics 131(4), 044108 (2009) PMid:19655838
View Article PubMed/NCBIYanai, T., Tew, D.P., Handy, N.C.: A New Hybrid Exchange-Correlation Functional Using the Coulomb-Attenuating Method (CAM-B3LYP). Chemical Physics Letters 393(1-3), 51–57 (2004)
View ArticleMarenich, A., Cramer, C., Truhlar, D.: Universal Solvation Model Based on Solute Electron Density and a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. Journal of Physical Chemistry B 113, 6378–6396 (2009) PMid:19366259
View Article PubMed/NCBIWeigend, F.: Accurate Coulomb-fitting Basis Sets for H to R. Physical Chemistry Chemical Physics 8, 1057–1065 (2006) PMid:16633586
View Article PubMed/NCBIWeigend, F., Ahlrichs, R.: Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Physical Chemistry Chemical Physics 7, 3297–3305 (2005) PMid:16240044
View Article PubMed/NCBIFrisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G.A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H.P., Izmaylov, A.F., Bloino, J., Zheng, G., Sonnenberg, J.L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery, J.A. Jr., Peralta, J.E., Ogliaro, F., Bearpark, M., Heyd, J.J., Brothers, E., Kudin, K.N., Staroverov, V.N., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A., Burant, J.C., Iyengar, S.S., Tomasi, J., Cossi, M., Rega, N., Millam, J.M., Klene, M., Knox, J.E., Cross, J.B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann: Gaussian 09 Revision E.01. Gaussian Inc., Wallingford CT, 2016
Domingo, L.R., Ríos-Gutiérrez, M., Pérez, P.: Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 21, 748 (2016) PMid:27294896 PMCid:PMC6273244
View Article PubMed/NCBIChamorro, E., Pérez, P., Domingo, L.R.: On the Nature of Parr Functions to Predict the Most Reactive Sites along Organic Polar Reactions. Chemical Physics Letters 582, 141–143 (2013)
View ArticleDomingo, L.R., Pérez, P., Sáez, J.A.: Understanding the Local Reactivity in Polar Organic Reactions through Electrophilic and Nucleophilic Parr Functions. RSC Advances 3, 1486–1494 (2013)
View ArticleMorell, C., Hocquet, A., Grand, A., Jamart-Grégoire, B.: A Conceptual DFT Study of Hydrazino Peptides: Assessment of the Nucleophilicity of the Nitrogen Atoms by Means of the Dual Descriptor ∆f(r). Journal of Molecular Structure: THEOCHEM 849, 46–51 (2008)
View ArticleMorell, C., Ayers, P.W., Grand, A., Gutiérrez–Oliva, S., Toro–Labbé, A.: Rationalization of the Diels-Alder Reactions through the Use of the Dual Reactivity Descriptor ∆f(r). Physical Chemistry Chemical Physics 10, 7239–7246 (2008) PMid:19060968
View Article PubMed/NCBIAyers, P.W., Morell, C., De Proft, F., Geerlings, P.: Understanding the Woodward-Hoffmann Rules by Using Changes in Electron Density. Chemistry - A European Journal 13, 8240–8247 (2007) PMid:17639522
View Article PubMed/NCBICárdenas, C., Rabi, N., Ayers, P.W., Morell, C., Jaramillo, P., Fuentealba, P.: Chemical Reactivity Descriptors for Ambiphilic Reagents: Dual Descriptor, Local Hypersoftness, and Electrostatic Potential. Journal of Physical Chemistry A 113, 8660–8667 (2009) PMid:19580251
View Article PubMed/NCBIMorell, C., Grand, A., Toro-Labbé, A.: Theoretical Support for Using the ∆f(r) Descriptor. Chemical Physics Letters 425, 342–346 (2006)
View ArticleMorell, C., Grand, A., Toro-Labbé, A.: New Dual Descriptor for Chemical Reactivity. Journal of Physical Chemistry A 109, 205–212 (2005) PMid:16839107
View Article PubMed/NCBIParr, R.G., Szentpaly, L.V., Liu, S.B.: Electrophilicity Index. Journal of the American Chemical Society 121, 1922–1924 (1999)
View ArticleParr, R.G., Yang, W.: Density Functional Approach to the Frontier-Electron Theory of Chemical Reactivity. Journal of the American Chemical Society 106, 4049–4050 (1984)
View ArticleJacquemin, D., Moore, B., Planchat, A., Adamo, C., Autschbach, J.: Performance of an Optimally Tuned Range-Separated Hybrid Functional for 0-0 Electronic Excitation Energies. Journal of Chemical Theory and Computation 10(4), 1677–1685 (2014) PMid:26580376
View Article PubMed/NCBICramer, C.J.: Essentials of Computational Chemistry - Theories and Models, 2nd edn. John Wiley & Sons, Chichester, England (2004)
Gledhill, J.D., De Proft, F., Tozer, D.J.: Range-Separation Parameter in Tuned Exchange-Correlation Functionals: Successive Ionizations and the Fukui Function. Journal of Chemical Theory and Computation 12(10), 4879–4884 (2016) PMid:27622316
View Article PubMed/NCBIJensen, F.: Introduction to Computational Chemistry, 2nd edn. John Wiley & Sons, Chichester, England (2007)
Lewars, E.: Computational Chemistry - Introduction to the Theory and Applications of Molecular and Quantum Mechanics. Kluwer Academic Publishers, Dordrecht (2003)
Young, D.C.: Computational Chemistry - A Practical Guide for Applying Techniques to Real-World Problems. John Wiley & Sons, New York (2001)
Easton, R.E., Giesen, D.J., Welch, A., Cramer, C.J., Truhlar, D.G.: The MIDI! Basis Set for Quantum Mechanical Calculations of Molecular Geometries and Partial Charges. Theoretical Chemistry Accounts 93, 281–301 (1996)
View ArticleHuzinaga, S., Andzelm, J., Klobukowski, M., Radzio-Audzelm, E., Sakai, Y., Tatewaki, H.: Gaussian Basis Sets for Molecular Calculations. Elsevier, Amsterdam (1984)
Shirahashi, Y., Watanabe, H., Hayase, F.: Identification of Red Pigments Formed in a D-Xylose-Glycine Reaction System. Bioscience, Biotechnology and Biochemistry 73(10), 2287–2292 (2009) PMid:19809196
View Article PubMed/NCBIToro–Labbé, A. (ed.): Theoretical Aspects of Chemical Reactivity. Elsevier Science, Amsterdam (2007)
Chattaraj, P.K. (ed.): Chemical Reactivity Theory - A Density Functional View. CRC Press. Taylor & Francis Group, Boca Raton (2009)
Parr, R.G., Yang, W.: Density-Functional Theory of Atoms and Molecules. Oxford University Press, New York (1989)
Geerlings, P., De Proft, F., Langenaeker, W.: Conceptual Density Functional Theory. Chemical Reviews 103, 1793–1873 (2003) PMid:12744694
View Article PubMed/NCBINursten, H. (ed.): The Maillard Reaction - Chemistry, Biochemistry and Implications. The Royal Society of Chemistry, Cambridge, UK (2005)
Frau, J., Flores-Holguín, N., Glossman-Mitnik, D.: Chemical Reactivity Properties, pKa Values, AGEs Inhibitor Abilities and Bioactivity Scores of the Mirabamides A–H Peptides of Marine Origin Studied by Means of Conceptual DFT. Marine Drugs 16(9), 302–19 (2018) PMid:30154377 PMCid:PMC6163382
View Article PubMed/NCBIFrau, J., Glossman-Mitnik, D.: Blue M2: An Intermediate Melanoidin Studied via Conceptual DFT. Journal of Molecular Modeling 24(138), 1–13 (2018)
View ArticleFrau, J., Glossman-Mitnik, D.: Chemical Reactivity Theory Applied to the Calculation of the Local Reactivity Descriptors of a Colored Maillard Reaction Product. Chemical Science International Journal 22(4), 1–14 (2018)
View ArticleFrau, J., Glossman-Mitnik, D.: Computational Study of the Chemical Reactivity of the Blue-M1 Intermediate Melanoidin. Computational and Theoretical Chemistry 1134, 22–29 (2018)
View ArticleFrau, J., Glossman-Mitnik, D.: Molecular Reactivity of some Maillard Reaction Products Studied through Conceptual DFT. Contemporary Chemistry 1(1), 1–14 (2018)
Frau, J., Glossman-Mitnik, D.: Conceptual DFT Study of the Local Chemical Reactivity of the Colored BISARG Melanoidin and Its Protonated Derivative. Frontiers in Chemistry 6(136), 1–9 (2018)
View ArticleFrau, J., Glossman-Mitnik, D.: Molecular Reactivity and Absorption Properties of Melanoidin Blue-G1 through Conceptual DFT. Molecules 23(3), 559–15 (2018) PMid:29498665 PMCid:PMC6017537
View Article PubMed/NCBIFrau, J., Glossman-Mitnik, D.: Conceptual DFT Study of the Local Chemical Reactivity of the Dilysyldipyrrolones A and B Intermediate Melanoidins. Theoretical Chemistry Accounts 137(5), 1210 (2018)
View ArticleArgirova, M.D.: Photosensitizer Activity of Model Melanoidins. Journal of Agricultural and Food Chemistry 53(4), 1210–1214 (2005) PMid:15713043
View Article PubMed/NCBIChai, J., Head-Gordon, M.: Systematic Optimization of Long-Range Corrected Hybrid Density Functionals. Journal of Chemical Physics 128, 084106 (2008) PMid:18315032
View Article PubMed/NCBIChai, J., Head-Gordon, M.: Long-Range Corrected Hybrid Density Functionals with Damped Atom-Atom Dispersion Corrections. Physical Chemistry Chemical Physics 10, 6615–6620 (2008) PMid:18989472
View Article PubMed/NCBIHalgren, T.A.: Merck Molecular Force Field. I. Basis, Form, Scope, Parameterization, and Performance of MMFF94. Journal of Computational Chemistry 17(5-6), 490–519 (1996) 1096-987X(199604)17:5/6<490::AID-JCC1>3.0.CO;2-P
View ArticleHalgren, T.A.: Merck Molecular Force Field. II. MMFF94 van der Waals and Electrostatic Parameters for Intermolecular Interactions. Journal of Computational Chemistry 17(5-6), 520–552 (1996) 1096-987X(199604)17:5/6<520::AID-JCC2>3.0.CO;2-W
View ArticleHalgren, T.A.: MMFF VI. MMFF94s Option for Energy Minimization Studies. Journal of Computational Chemistry 20(7), 720–729 (1999) 1096-987X(199905)20:7<720::AID-JCC7>3.0.CO;2-X
View ArticleHalgren, T.A., Nachbar, R.B.: Merck Molecular Force Field. IV. Conformational Energies and Geometries for MMFF94. Journal of Computational Chemistry 17(5-6), 587–615 (1996) 1096-987X(199604)17:5/6<587::AID-JCC4>3.0.CO;2-Q
View ArticleHalgren, T.A.: Merck Molecular Force field. V. Extension of MMFF94 Using Experimental Data, Additional Computational Data, and Empirical Rules. Journal of Computational Chemistry 17(5-6), 616–641 (1996) 1096-987X(199604)17:5/6<616::AID-JCC5>3.0.CO;2-X
View ArticleEgger, D.A., Weissman, S., Refaely-Abramson, S., Sharifzadeh, S., Dauth, M., Baer, R., Ku ̈mmel, S., Neaton, J.B., Zojer, E., Kronik, L.: Outer-Valence Electron Spectra of Prototypical Aromatic Heterocycles From an Optimally Tuned Range-Separated Hybrid Functional. Journal of Chemical Theory and Computation 10(5), 1934–1952 (2014) PMid:24839410 PMCid:PMC4020925
View Article PubMed/NCBIFoster, M.E., Wong, B.M.: Nonempirically Tuned Range-Separated DFT Accurately Predicts Both Fundamental and Excitation Gaps in DNA and RNA Nucleobases. Journal of Chemical Theory and Computation 8(8), 2682–2687 (2012) PMid:22904693 PMCid:PMC3419459
View Article PubMed/NCBIFoster, M.E., Azoulay, J.D., Wong, B.M., Allendorf, M.D.: Novel Metal–Organic Framework Linkers for Light Harvesting Applications. Chemical Science 5(5), 2081–2090 (2014)
View ArticleKarolewski, A., Stein, T., Baer, R., Kummel, S.: Communication: Tailoring the Optical Gap in Light-Harvesting Molecules. The Journal of Chemical Physics 134(15), 151101–5 (2011) PMid:21513368
View Article PubMed/NCBIKarolewski, A., Kronik, L., Kummel, S.: Using Optimally Tuned Range Separated Hybrid Functionals in Ground-State Calculations: Consequences and Caveats. The Journal of Chemical Physics 138(20), 204115 (2013) PMid:23742462
View Article PubMed/NCBIKoppen, J.V., Hapka, M., Szczeniak, M.M., Chalasinski, G.: Optical Absorption Spectra of Gold Clusters Au(n) (n = 4, 6, 8,12, 20) From Long-Range Corrected Functionals with Optimal Tuning. The Journal of Chemical Physics 137(11), 114302 (2012) PMid:22998257
View Article PubMed/NCBIKronik, L., Stein, T., Refaely-Abramson, S., Baer, R.: Excitation Gaps of Finite-Sized Systems from Optimally Tuned Range-Separated Hybrid Functionals. Journal of Chemical Theory and Computation 8(5), 1515–1531 (2012) PMid:26593646
View Article PubMed/NCBIKuritz, N., Stein, T., Baer, R., Kronik, L.: Charge-Transfer-Like π → π* Excitations in Time-Dependent Density Functional Theory: A Conundrum and Its Solution. Journal of Chemical Theory and Computation 7(8), 2408–2415 (2011) PMid:26606616
View Article PubMed/NCBILima, I.T., Prado, A.d.S., Martins, J.B.L., de Oliveira Neto, P.H., Ceschin, A.M., da Cunha, W.F., da Silva Filho, D.A.: Improving the Description of the Optical Properties of Carotenoids by Tuning the Long-Range Corrected Functionals. The Journal of Physical Chemistry A 120(27), 4944–4950 (2016) PMid:26885879
View Article PubMed/NCBIManna, A.K., Lee, M.H., McMahon, K.L., Dunietz, B.D.: Calculating High Energy Charge Transfer States Using Optimally Tuned Range-Separated Hybrid Functionals. Journal of Chemical Theory and Computation 11(3), 1110–1117 (2015) PMid:26579761
View Article PubMed/NCBIMoore II, B., Autschbach, J.: Longest-Wavelength Electronic Excitations of Linear Cyanines: The Role of Electron Delocalization and of Approximations in Time-Dependent Density Functional Theory. Journal of Chemical Theory and Computation 9(11), 4991–5003 (2013) PMid:26583416
View Article PubMed/NCBINiskanen, M., Hukka, T.I.: Modeling of Photoactive Conjugated Donor-Acceptor Copolymers: the Effect of the Exact HF Exchange in DFT Functionals on Geometries and Gap Energies of Oligomer and Periodic Models. Phys. Chem. Chem. Phys. 16(26), 13294–13305 (2014)
View ArticlePereira, T.L., Leal, L.A., da Cunha, W.F., Timóteo de Sousa Júnior, R., Ribeiro Junior, L.A., Antonio da Silva Filho, D.: Optimally Tuned Functionals Improving the Description of Optical and Electronic Properties of the Phthalocyanine Molecule. Journal of Molecular Modeling 23(3), 71 (2017) PMid:28197842
View Article PubMed/NCBIPhillips, H., Zheng, S., Hyla, A., Laine, R., Goodson III, T., Geva, E., Dunietz, B.D.: Ab Initio Calculation of the Electronic Absorption of Functionalized Octahedral Silsesquioxanes via Time-Dependent Density Functional Theory with Range-Separated Hybrid Functionals. The Journal of Physical Chemistry A 116(4), 1137–1145 (2012) PMid:22191709
View Article PubMed/NCBIPhillips, H., Geva, E., Dunietz, B.D.: Calculating Off-Site Excitations in Symmetric Donor-Acceptor Systems via Time-Dependent Density Functional Theory with Range-Separated Density Functionals. Journal of Chemical Theory and Computation 8(8), 2661–2668 (2012) PMid:26592111
View Article PubMed/NCBIRefaely-Abramson, S., Baer, R., Kronik, L.: Fundamental and Excitation Gaps in Molecules of Relevance for Organic Photovoltaics From an Optimally Tuned Range-Separated Hybrid Functional. Physical Review B 84(7), 075144–8 (2011)
View ArticleStein, T., Kronik, L., Baer, R.: Prediction of Charge-Transfer Excitations in Coumarin-Based Dyes Using a Range-Separated Functional Tuned From First Principles. The Journal of Chemical Physics 131(24), 244119 (2009) PMid:20059066
View Article PubMed/NCBIStein, T., Kronik, L., Baer, R.: Reliable Prediction of Charge Transfer Excitations in Molecular Complexes Using Time-Dependent Density Functional Theory. Journal of the American Chemical Society 131(8), 2818–2820 (2009) PMid:19239266
View Article PubMed/NCBISun, H., Autschbach, J.: Electronic Energy Gaps for π-Conjugated Oligomers and Polymers Calculated with Density Functional Theory. Journal of Chemical Theory and Computation 10(3), 1035–1047 (2014) PMid:26580181
View Article PubMed/NCBIBecke, A.D.: Vertical Excitation Energies From the Adiabatic Connection. The Journal of Chemical Physics 145(19), 194107 (2016) PMid:27875864
View Article PubMed/NCBIBaerends, E.J., Gritsenko, O.V., van Meer, R.: The Kohn-Sham Gap, the Fundamental Gap and the Optical Gap: The Physical Meaning of Occupied and Virtual Kohn-Sham Orbital Energies. Physical Chemistry Chemical Physics 15(39), 16408–16425 (2013) PMid:24002107
View Article PubMed/NCBIvan Meer, R., Gritsenko, O.V., Baerends, E.J.: Physical Meaning of Virtual Kohn-Sham Orbitals and Orbital Energies: An Ideal Basis for the Description of Molecular Excitations. Journal of Chemical Theory and Computation 10(10), 4432–4441 (2014) PMid:26588140
View Article PubMed/NCBIGázquez, J.L., Cedillo, A., Vela, A.: Electrodonating and Electroaccepting Powers. Journal of Physical Chemistry A 111(10), 1966–1970 (2007) PMid:17305319
View Article PubMed/NCBIChattaraj, P.K., Chakraborty, A., Giri, S.: Net Electrophilicity. Journal of Physical Chemistry A 113(37), 10068–10074 (2009) PMid:19702288
View Article PubMed/NCBIZhurko, G.A., Zhurko, D.A.: Chemcraft program Revision 1.6. Grigoriy A. Zhurko, United States (2012).
View Article