Aliaksandr V. Faryna
Email: farina@iboch.by
© 2019 Sift Desk Journals. All Rights Reserved
VOLUME: 3 ISSUE: 2
Page No: 285-293
Aliaksandr V. Faryna
Email: farina@iboch.by
Aliaksandr V. Faryna*, Еlena N. Kalinichenko
Institute of Bioorganic Chemistry, National Academy of Sciences of Belarus, BY-220141, 5/2 Academician V.F. Kuprevich Street, Minsk, Republic of Belarus
Xin Wang(wx8399@ouc.edu.cn)
Zhenming Liu(zmliu@bjmu.edu.cn)
A. Faryna,Е. N. Kalinichenko, Computer-aided molecular design of new potential inhibitors of protein kinases using of 4-methyl-benzoic acid as a linker(2019)Journal of Computational Chemistry & Molecular Modeling 3(2)pp:285-293
Considering pharmacophore features of known protein kinase inhibitors we created virtual library of new 4-methylbenzamide derivatives. Docking of target structures was performed using nine cancer-related protein kinases as receptors. Docking protocol was validated using approved kinase inhibitors. Protein-ligand complexes with the best score were treated by visual inspection and the structure 7 containing benzimidazole k for ATP-site and fragment of 3-(4-methyl-1H-imidazole-1-yl)-5-(trifluoromethyl)aniline f was considered as the most promising substance. Based on docking results, the most favorable structures were evaluated via molecular dynamics with MM/PBSA free binding energy calculation showing in some cases comparable with known inhibitors results in terms of total binding energy, polar and non-polar interactions with best result -160.0 kJ/mol for T315I-mutant ABL kinase. Mean binding energy for approved kinase inhibitors was -161.0 kJ/mol. It was shown, that directed structural modifications of initial structures could further increase calculated binding energy. The combined docking and molecular dynamics results suggest that proposed structures could be valuable objects in the development of new kinase inhibitors with the derivatives of 4-[(1H-1,3,-benzodiazole-2-yl)methyl]-N-phenylbenzamide being most promising ones having docking score of -12.6 and binding energy of -157.6 kJ/mol in the respect of human ABL kinase.
Keywords: Protein kinase inhibitors, virtual screening, molecular dynamics, PDB-complex, 4-methylbenzamide derivatives
Targeted therapy is a modern approach for the treatment of various cancer diseases, which aims to treat cancer specific biological targets [1]. Small-molecule protein kinase inhibitors (PKIs) constitute the essential part of targeted therapy options with more than 30 drugs approved so far [2]. Kinase activity in cancer cells is often deregulated altering cell signaling pathways thus increasing cell proliferation. PKIs selectively bind to the active site of a cancer-related protein kinase and suppress its biological functions. PKIs have shown great effectiveness in cancer treatment, but their use is often limited due to serious side effects as well as several types of resistance including point mutations of original kinase [3-5]. Hence, new kinase inhibitors that are more selective and can override current limitations are of great need.
Molecular docking and molecular dynamics have become common tools in structure-based drug design [6-8]. Docking is generally used for structure modeling of protein-ligand complexes along with rough estimation of ligand binding affinity. Docking results can also explain biological activity of known inhibitors. Docking is also a useful tool for primary virtual screening of large virtual libraries of chemical structures. Molecular dynamics studies of protein-ligand complexes are widely used for time-dependent stability studies and more accurate binding energy calculation.
In this work new derivatives of 4-methyl benzoic acid amides have been evaluated in silico as kinase inhibitors using combined molecular docking and molecular dynamics approach.
2.1. Design of target structures
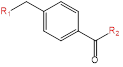
Combinatory method was used to create library of target structures starting from 4-methyl-benzamide (Fig. 1). Molecule of nilotinib was used for modeling as a reference structure. Nilotinib is a second-generation tyrosine kinase inhibitor which targets BCR-ABL tyrosine kinase in inactive conformation. Being a type II inhibitor nilotinib exploits inactive kinase conformation to get additional interactions in the allosteric pocket of kinase binding site. Hence, the structure of nilotinib could be divided into three regions: ATP-site fragment, allosteric fragment and hydrophobic linker (Fig. 2) [9].
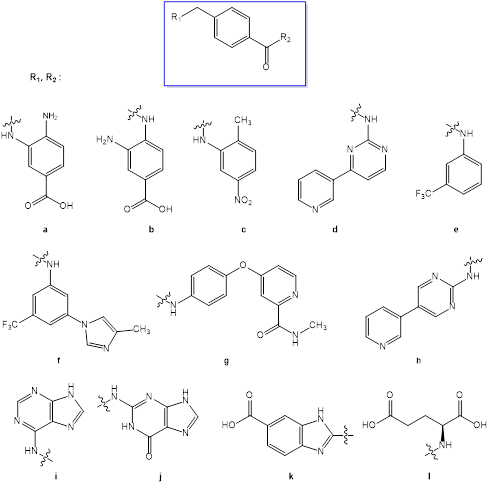
Figure 1: Generation of target structures
Target structures were design in the way to maintain the main pharmacophore features of nilotinib: amide bond for allosteric site interactions as well as heterocycle aromatic system for π-π stacking and hydrogen bonds formation in ATP site. The key difference of target structures compared to niltotinib is utilizing p-position to come out from benzamide linker into ATP-binding site instead of m-position in the case of nilotinib. Similar linker choice could be seen in modern protein kinas inhibitors such as sorafenib and rebastinib (Fig. 2). On the other hand, binding sites of kinases are known to have a lot of similarities between different classes of the enzymes. That’s why we consider proposed linker modification to be promising for discovering new kinase inhibitors.
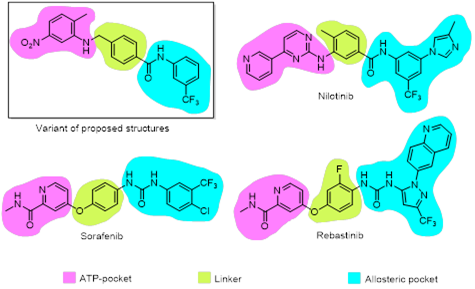
Figure 2: Design approach summary
2.2. Docking
Docking was implemented using AutoDock Vina software [10]. Forty complexes of different protein kinases with approved kinase inhibitors were chosen from Protein Data Bank (PDB). Complexes were filtered to give only nine complexes of type II inhibitors [11]. Proteins were extracted and used as receptors for docking. The structures of these receptors were minimized to give nine additional receptors (Tab. 1). Preliminary, the docking validation was performed using original ligands of PDB-complexes [12]. Ligand structures were extracted and minimized using two different force fields: Merck Molecular Force Field (MMFF94s) and Generalized Amber Force Field (GAFF). Minimization of original ligands was performed to imitate their construction from scratch without knowing real binding mode. As a result, three ligand structures were obtained for each PDB-record. Docking results of these ligands were further used as binding affinity threshold to filter target structures. Further, for each target structure three geometric conformers were generated considering that AutoDock Vina is not able to change any valent angles in ligand during the docking. All final conformers were docked and docking results were filtered to give protein-ligand complexes for further molecular dynamics studies.
All chemical structure file format conversions were done using Open Babel [13]. Ligands from PDB-complexes were extracted by copying of HEATATM section of pdb-file. Two-dimensional structures were created with Marvin Sketch [14]. 3D-structures were generated using Molconvert [15]. Conformations generation and structure minimization were performed using Open Babel genetic algorithm. Missing protein residues were restored with MODELLER [16]. Water and ions were removed from receptors. GROMACS with AMBER FF99SB-ILDN force field was used for obtaining minimized receptors. Binding site coordinates of receptors were determined by the size of an original ligand in PDB-file and were scaled-up by 20% in each dimension. Docking results were limited by only one docking pose with the best docking score. Preparation of ligands and receptors for docking was carried out in MGL Tools [17]. Visualization of docking results was made in Chimera [18].
Table 1: PDB-complexes of type II kinase inhibitors used for docking and binding energy calculations along with the docking scores and RMSD values obtained for the original ligands
|
PDB record |
Receptor type/Original ligand |
Docking score for non-minimized receptors |
RMSDa for non-minimized receptors, Å |
Docking score for minimized receptors |
||||||
|
Non-min.b |
MMFF94s |
GAFF |
Non min.b |
MMFF94s |
GAFF |
Non-min.b |
MMFF94s |
GAFF |
||
|
2HYY |
ABL/imatinib |
-12,7 |
-10,2 |
-12,8 |
0,96 |
1,69 |
0,48 |
-12,5 |
-12,5 |
-12,6 |
|
2PL0 |
LCK/imatinib |
-10,6 |
-8,7 |
-11,5 |
1,67 |
12,73 |
0,94 |
-10,6 |
-9,4 |
-9,0 |
|
3CS9 |
ABL/nilotinib |
-13,6 |
-13,6 |
-13,8 |
0,95 |
0,95 |
0,96 |
-11,6 |
-11,5 |
-11,7 |
|
3GCS |
P38MAP/sorafenib |
-10,7 |
-10,7 |
-10,7 |
1,83 |
1,79 |
1,85 |
-10,9 |
-10,9 |
-10,9 |
|
3WZE |
KDR/sorafenib |
-12,3 |
-12,2 |
-12,3 |
0,79 |
0,83 |
0,82 |
-11,5 |
-11,4 |
-11,6 |
|
3QRJ |
ABL/rebastinib |
-14,5 |
-14,3 |
-13,2 |
1,47 |
1,47 |
0,63 |
-11,9 |
-12,1 |
-10,8 |
|
4AG8 |
VEGFR2/axitinib |
-10,7 |
-10,2 |
-10,4 |
0,65 |
0,69 |
1,48 |
-10,0 |
-9,8 |
-9,8 |
|
4ASD |
VEGFR2/sorafenib |
-11,8 |
-11,6 |
-11,8 |
1,27 |
0,92 |
1,26 |
-11,6 |
-11,5 |
-11,6 |
|
5HI2 |
BRAF/sorafenib |
-11,2 |
-11,2 |
-11,1 |
0,98 |
0,84 |
1,18 |
-11,0 |
-11,0 |
-10,8 |
aRMSD – root mean squared deviation of heavy atoms
bnon-minimized structure
2.3. Molecular dynamics
GROMACS was used for molecular dynamics simulation [19]. General simulation workflow included energy minimization step, two equilibration steps and 2 ns production run. Final MD trajectory was snapshotted and binding energy was calculated using the method of molecular mechanics energies combined with the Poisson–Boltzmann surface area (MM/PBSA). G_mmpbsa software was used for energy calculation [20].
AMBER FF99SB-ILDN force field was used for simulation. All ligands were parametrized with ACPYPE [13]. Simulation workflow was as follows: solvation, neutralizing and adding NaCl ions up to concentration of 0.15 mol, energy minimization, NPT and NVT equilibration steps 200 ps each and final 2 ns production run. Dodecahedron box of 1.2 nm and periodic bounding conditions were used. Berendsen thermostat was used for equilibration. Long range electrostatic interactions were treated according to PME method. Every twentieth frame of final molecular dynamics trajectory was extracted skipping first 100 frames to perform energy calculations.
Docking results of original ligands with minimized and non-minimized receptors are given in Tab. (1). For non-minimized receptors, in the most cases, the final docking poses of original ligands matched experimental data with root mean squared deviation (RMSD) less than 2.0 Å. Our docking protocol was not able to reproduce experimental binding pose of imatinib in 2PL0 complex when ligand was minimized using MMFF94s force field (RMSD 12.73 Å). For minimized receptors the validation of docking results of original ligands was performed by visual inspection and all obtained docking poses were considered to be valid in terms of ligand placement. Mean docking score of valid docking poses of original inhibitors was of -11.9 for non-minimized receptors and -11.1 for minimized ones. GAFF force filed was used for further processing of target structures and docking score of -11.5 was considered as a threshold for filtering results.
According to proposed combinatorial method, 144 target structures were generated in three different conformations, totally 432 conformations. Each conformation was GAFF-minimized and docked into both minimized and non-minimized receptors (Fig. 3).
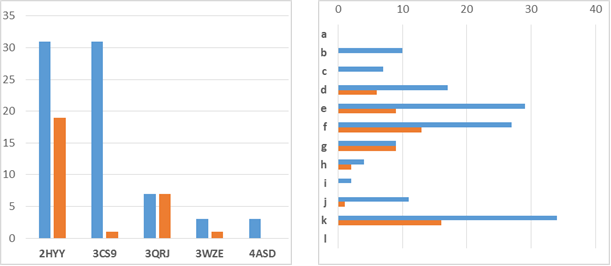
Figure 3: The number of conformations with docking score better than -11.5 and number of appearance of different structural fragments in them for minimized (red) and non-minimized (blue) receptors
In the case of non-minimized receptors, total number of conformations with the score better than -11.5 was 75, which corresponded to 50 unique ligand structures. For minimized receptors this docking score showed 28 conformations and 18 unique structures. Benzimidazole k was the most common structural fragment for above-mentioned poses (Fig. 3). Fourteen ligand structures showed docking score better than -11.5 for both minimized and non-minimized receptors. Thus, the obtained results suggested that pharmacophore features of proposed ligand structures are in general favorable for the placement in the binding site of a kinase. Protein-ligand complexes with the best score were treated by visual inspection and the structure 7 containing benzimidazole k for ATP-site and fragment of 3-(4-methyl-1H-imidazole-1-yl)-5-(trifluoromethyl)aniline f was considered as the most promising substance. This complex is characterized by high docking scores: -12.6 for non-minimized C-ABL (PDB 2HYY) and -12.3 for non-minimized Human ABL (PDB 3CS9). The best binding poses, obtained by AutoDock Vina, were in the agreement with the initial design logic of target structures. Docking results of the structure 7 showed typical for type II inhibitors binding characteristics. Trifluoromethylaniline residues occupied the allosteric pocket, and the position of amide bond was similar to those of known inhibitors. Benazamide linker with rotatable methylene group made it possible for benzimidazole moiety to be placed in ATP-pocket of binding site. Further analysis of interactions showed the presence of hydrogen bonds with amino acid residues Met-318, Thr-315, Asp-381, Glu-286 and Thr-251 (Fig. 4).
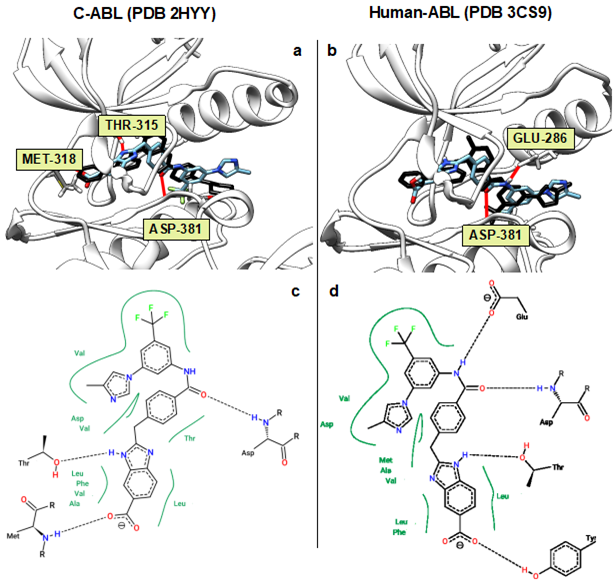
Figure 4: (a) Superimposed binding poses of imatinib (experimental, black) and structure of 7 (AutoDock Vina), C-ABL kinase (PDB 2HYY). (b) Superimposed binding poses of nilotinib (experimental, black) and structure 7 (AutoDock Vina), Human ABL kinase (PDB 3CS9). (c) Interactions map of structure 7 and C-ABL. (d) Interactions map of structure 7 and Human ABL. Hydrogen bonds for 3D-structures are predicted with Chimera [20] and highlighted with red. PoseView [21] was used to generate 2D-interactions maps
Complexes of target structures with best docking scores as well as some other complexes showing adequate ligand placement together with complexes of original ligands were processed to molecular dynamics step. Sixteen complexes of target structures and nine complexes of original ligands were taken to the molecular dynamics studies for free binding energy calculations using MM/PBSA method. Mean calculated binding energy for original and studied ligands was equal to -161.0 kJ/mol and -121.3 kJ/mol respectively (Tab. 2).
Table 2: Calculated binding affinities and their decompositions (kJ/mol) for complexes of target structures, references and modifications
|
|
||||||||
|
Structure |
R1 |
R2 |
PDB record |
Docking score |
VdW |
Electrostatic |
Polar solvation |
Binding energy |
|
1 |
e |
h |
2HYY |
-11.9 |
-226.7+/-12.5 |
-28.4+/-9.4 |
169.6+/-19.3 |
-108.2+/-16.5 |
|
2 |
e |
f |
2HYY |
-11.6 |
-261.1+/-11.6 |
-38.4+/-10.2 |
187.6+/-14.8 |
-137.6+/-13.9 |
|
2 |
e |
f |
3qrj |
-12.2 |
-261.2+/-14.4 |
-20.7+/-7.6 |
168.1+/-17.1 |
-139.2+/-15.6 |
|
3 |
k |
e |
2HYY |
-12.2 |
-238.6+/-8.5 |
-46.9+/-10.0 |
207.6+/-15.7 |
-101.2+/-15.2 |
|
3 |
k |
e |
5HI2 |
-11.4 |
-222.9+/-10.8 |
-65.6+/-9.4 |
204.0+/-11.6 |
-107.5+/-13.1 |
|
4 |
c |
f |
2HYY |
-11.4 |
-270.9+/-10.7 |
-68.5+/-11.1 |
222.2+/-14.4 |
-144.1+/-12.5 |
|
5 |
g |
f |
3qrj |
-12.3 |
-289.1+/-11.8 |
-18.2+/-14.1 |
175.7+/-21.0 |
-160.0+/-13.7 |
|
5 |
g |
f |
2HYY |
-8.9 |
-249.7+/-18.7 |
-75.8+/-19.3 |
228.0+/-42.7 |
-123.4+/-31.0 |
|
6 |
b |
f |
3qrj |
-11.4 |
-257.9+/-12.6 |
-13.4+/-9.6 |
183.3+/-17.6 |
-113.9+/-22.8 |
|
6 |
b |
f |
3cs9 |
-11.8 |
-252.2+/-12.1 |
-77.6+/-12.1 |
217.8+/-18.3 |
-137.8+/-11.3 |
|
7 |
k |
f |
3cs9 |
-12.3 |
-273.0+/-13.6 |
-64.3+/-18.6 |
255.1+/-26.7 |
-109.3+/-18.3 |
|
8 |
k |
d |
3cs9 |
-12.5 |
-255.9+/-11.5 |
-45.9+/-11.9 |
214.6+/-16.2 |
-110.7+/-14.3 |
|
9 |
f |
k |
3cs9 |
-12.2 |
-279.3+/-11.9 |
-43.8+/-14.8 |
251.1+/-24.1 |
-99.6+/-17.2 |
|
10 |
d |
k |
3cs9 |
-12.1 |
-234.2+/-12.7 |
-35.1+/-11.7 |
210.0+/-19.6 |
-82.3+/-16.1 |
|
11 |
g |
b |
3cs9 |
-11.4 |
-271.9+/-14.5 |
-52.3+/-15.8 |
222.7+/-15.8 |
-128.6+/-28.3 |
|
12 |
d |
f |
3qrj |
-11.7 |
-284.7+/-12.8 |
-35.6+/-10.5 |
210.5+/-21.6 |
-137.1+/-14.0 |
|
imatinib |
- |
- |
2HYY |
-12.8 |
-282.1+/-10.0 |
-42.4+/-7.4 |
195.6+/-10.4 |
-154.7+/-12.3 |
|
imatinib |
- |
- |
2PL0 |
-11.5 |
-255.5+/-13.2 |
-36.5+/-10.2 |
171.5+/-12.3 |
-146.0+/-12.3 |
|
sorafenib |
- |
- |
5HI2 |
-11.1 |
-243.3+/-13.1 |
-81.4+/-9.3 |
210.1+/-9.5 |
-137.9+/-13.1 |
|
sorafenib |
- |
- |
3WZE |
-12.3 |
-254.8+/-8.1 |
-73.4+/-8.3 |
188.7+/-8.4 |
-162.8+/-11.6 |
|
ponatinib |
- |
- |
2HYY |
-11.6 |
-290.9+/-10.9 |
-32.9+/-8.7 |
191.0+/-20.0 |
-159.4+/-18.4 |
|
ponatinib |
- |
- |
3cs9 |
-11.6 |
-273.2+/-13.1 |
-47.7+/-15.2 |
198.4+/-24.1 |
-149.3+/-20.2 |
|
nilotinib |
- |
- |
3cs9 |
-13.8 |
-304.1+/-12.4 |
-63.2+/-10.6 |
216.4+/-13.2 |
-177.7+/-11.6 |
|
rebastinib |
- |
- |
3qrj |
-13.2 |
-314.4+/-10.8 |
-66.7+/-7.8 |
224.2+/-11.8 |
-185.3+/-12.8 |
|
M1 |
- |
- |
3cs9 |
-12.1 |
-301.2+/-11.2 |
-73.3+/-10.1 |
253.9+/-12.6 |
-148.0+/-11.2 |
|
M2 |
- |
- |
3cs9 |
-11.8 |
-263.4+/-8.6 |
-56.0+/-11.6 |
211.9+/-13.5 |
-132.6+/-12.7 |
|
M3 |
- |
- |
3cs9 |
-12.5 |
-291.0+/-11.9 |
-60.8+/-9.1 |
221.8+/-15.6 |
-157.6+/-12.4 |
|
M4 |
- |
- |
3cs9 |
-11.6 |
-273.0+/-11.7 |
-67.3+/-10.7 |
224.2+/-14.9 |
-141.7+/-15.1 |
|
M5 |
- |
- |
3cs9 |
-10.8 |
-248.0+/-12.9 |
-19.0+/-11.6 |
181.0+/-20.8 |
-110.0+/-20.8 |
Structure 5 showed comparable with known inhibitors result in the case of mutant T315I ABL-kinase and with binding energy equal to -160 kJ/mol. We performed binding energy decomposition and some structures showed comparable with known inhibitors values of polar and non-polar components. In many cases, total value of free binding energy of target structures was lowered by polar solvation energy (Fig. 5).
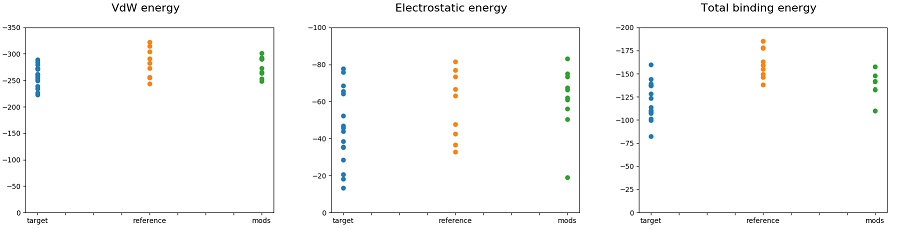
Figure 5: Calculated binding energies and their decomposition to non-polar and polar components of initial ligands, reference ligands and modifications
Structure 7 was the most promising one after docking step but showed poor total binding affinity but relatively high polar and non-polar components. That’s why we made attempts to increase estimated binding affinity of studied ligands by further structural modifications. Four modifications of structure 7 (M1-M4) and one modification of structure 4 (M5) were proposed and evaluated by the above-described workflow starting from obtaining docking pose followed by molecular dynamics simulation (Fig. 6).
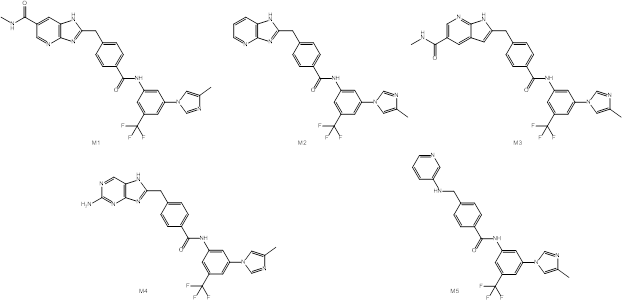
Figure 6: Proposed modifications of structures 4 and 7
For the modification of structure 4 (M5) the decrease in calculated binding affinity was observed, while modifications of structure 7 showed great improvement in calculated binding energy with best value of -157.6 kJ/mol for modification M3 (Tab. 2).
As a result of the study, the docking of the combinatorial library of 144 unique structures of type II protein kinase inhibitor analogues in three different conformations was implemented. The proposed approach to the design of the new compounds was aimed to increase the overall spatial extent of the linker as a result of the additional introduction of the methylene group into the benzene ring of the target structure. Under the described conditions of the inhibitory activity evaluation in silico fourteen ligand structures showed a better docking result than -11.5, both for non-minimized receptors and minimized ones. Structure 7, probably, appears to bind in the active kinase center as a type II inhibitor. In this case, the remainder of trifluoromethylaniline is located in the allosteric site of the active center, and the position of the amide group is similar to that of known inhibitors. Benzamide linker with mobile CH2 group makes it possible a favorable location of the benzimidazole fragment in the ATP binding site. This complex is characterized by high docking scores equal to -12.6 (non-minimized receptor) and -12.0 (minimized one) with ABL-kinase (2HYY) along with the presence of hydrogen bonds with amino acid residues Met-318, Thr-315 and Asp-381. It is shown that further modification of the basic structure 7 can increase the calculated binding energy. High value of binding efficiency for many candidates allows us to consider them as promising objects for further study by other computer methods, as well as for chemical synthesis and investigations of their biological properties in order to create new protein kinase inhibitors.
The authors are grateful for the financial support from the National Academy of Sciences of Belarus within the research project number 5.20 of State Program “Chemical Technologies and Materials”, subprogram 5 “Pharmacology and Pharmacy”.
Urruticoechea A, Alemany R, Balart J, Villanueva A, Viñals F, Capellá G (2010) Recent advances in cancer therapy: an overview. Curr Pharm Des 16(1):3-10.
View ArticleBhullar KS, Lagarón NO, Mcgowan EM et al (2018) Kinase-targeted cancer therapies: progress, challenges and future directions. Mol Cancer 17(1):48.
View ArticleRossari F, Minutolo F, Orciuolo E (2018) Past, present, and future of Bcr-Abl inhibitors: from chemical development to clinical efficacy. J Hematol Oncol 11(1):84.
View ArticleApperley JF (2007) Part I: mechanisms of resistance to imatinib in chronic myeloid leukaemia. Lancet Oncol 8:1018–1029. 70342-x
View ArticleRossari F, Minutolo F, Orciuolo E (2018) Past, present, and future of Bcr-Abl inhibitors: from chemical development to clinical efficacy. J Hematol Oncol 11(1):84.
View ArticleAci-sèche S, Ziada S, Braka A, Arora R, Bonnet P (2016) Advanced molecular dynamics simulation methods for kinase drug discovery. Future Med Chem 8(5):545-66.
View ArticleSliwoski G, Kothiwale S, Meiler J, Lowe EW (2014) Computational Methods in Drug Discovery. Pharmacological Reviews 66:334‐395.
View ArticleŚledź P, Caflisch A (2018) Protein Structure‐Based Drug Design: From Docking to Molecular Dynamics. Current opinion in structural biology 48:93‐102.
View ArticleRoskoski R Jr (2016) Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacological Reviews 103:26-48.
View ArticleTrott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. Journal of Computational Chemistry 31:455-461.
View ArticleVijayan, RSK, He P, Modi V, Duong-Ly KC, Ma H, Peterson JR, Dunbrack Jr RL, Levy RM (2015) Conformational Analysis of the DFG-Out Kinase Motif and Biochemical Profiling of Structurally Validated Type II Inhibitors. J Med Chem 58:466−479.
View ArticleBerman HM et al (2000) The Protein Data Bank. Nucleic Acids Research 28(1):235-242.
View ArticleAbraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E (2015) GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1:19-25.
View ArticleKumari R, Kumar R, Lynn A (2014) g_mmpbsa--a GROMACS tool for high-throughput MM-PBSA calculations. J Chem Inf Model 54(7):1951-62.
View ArticleO'Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR (2011) Open Babel: An open chemical toolbox. Journal of cheminformatics 3(1):33.
View ArticleMarvin 17.21.0, ChemAxon.
View ArticleMolecule file conversion with MolConverter.
View ArticleWebb B, Sali A (2014) Comparative protein structure modeling using MODELLER. Current protocols in bioinformatics 47(1):5.6.1-5.6.37.
View ArticleMorris GM, Huey R, Lindstrom W, Sanner M, Belew RK, Goodsell DS, Olson AJ (2009) AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of computational chemistry 30(16):2785-2791.
View ArticlePettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera—a visualization system for exploratory research and analysis. Journal of computational chemistry 25(13):1605-1612.
View ArticleStierand K, Matthias R (2010) Drawing the PDB: Protein−Ligand Complexes in Two Dimensions. ACS Medicinal Chemistry Letters 1(9):540-545.
View Article