Mr. Sajjad Esmaeili
E-mail: sajad.smaeili@kums.ac.ir
© 2019 Sift Desk Journals. All Rights Reserved
VOLUME: 4 ISSUE: 3
Page No: 440-451
Mr. Sajjad Esmaeili
E-mail: sajad.smaeili@kums.ac.ir
Yazdan Rahmati1, Soodabeh Shafiee1,2, Hasan Mollanoori1, Sajjad Esmaeili3*
1 Department of Medical Genetics, Iran University of Medical Sciences, Tehran, Iran
2 Barati medical diagnostic laboratory, Gachsaran, Iran
3 Medical Biology Research Center, Health Technology Institute, Kermanshah University of Medical Sciences, Kermanshah, Iran
Y. Rahmati, S. Shafiee, H. Mollanoori, S. Esmaeili, Contribution of commensal gut microbial in Kawasaki disease pathogenesis by weighted gene co-expression network analysis(2020)Journal of Computational Chemistry & Molecular Modeling 4(3)
Kawasaki disease (KD) is an acute and self-limited vasculitis which has unknown etiology with little described genetic mechanism. To achieve further insight into the molecular mechanisms underlying KD and identify new therapeutic targets for acute KD, we compared the acute and convalescent KD transcriptional profiles by bioinformatics tools. Recently, a novel co-expression algorithm, weighted gene co-expression network analysis (WGCNA), has provided more comprehensive data analysis. In the present study, the WGCNA package was applied to the datasets of KD patients obtained from the Gene Expression Omnibus (GEO). We identified network modules of co-expressed genes in the acute phase subject dataset and verified their preservation in the convalescent dataset. Then, in the non-preserved modules, we selected hub genes that distinguishing acute from convalescent phase and performed functional enrichment analyses of genes involved in these modules. We identified several genes that may play essential role in KD pathogenesis and discriminating acute from convalescent phase patients. Functional enrichment analysis revealed that most of them contribute to innate immune system defense against infectious agents. Neutrophil mediated immunity by antimicrobial humoral response against infectious agents. In conclusion our results indicated several genes essential for KD pathogenesis using WGCNA algorithm. These genes may be used as potential novel therapeutic targets.
Keywords: Kawasaki disease, WGCNA, innate immune response, antimicrobial peptides, acute phase, convalescent phase
KD is an inflammatory vasculitis with unknown etiology that occurs in infantile and toddlers. Its main complication is the development of coronary artery lesions (CALs) including coronary arterial dilation, stenosis, and aneurysms [1-3]. Kawasaki disease (KD) exhibits many signs such as high temperature, erythema which occurs in the mucosa of lips or mouth, alterations in the body organs, and large lymph nodes in the neck [4-6]. KD among Japanese and Japanese American children is clearly more common (265/100,000) than other populations like other Asian countries (51 to 194/100,000), Europe (8.39/100,000) or American children (20.8/100,000) [7-12]. To date, the etiology of KD has remained an undetermined question and the causative agents are also unexplained [13]. However, based on research studies, genetic predisposition considered a cause for the development of KD and also the interaction with an unknown infectious source can predispose to this disease [14]. The observation of familial occurrence and raised frequency in Asian populations, taken together suggest the existence of a genetic basis [15, 16]. Associations with genetic variants in several genes such as BLK, CASP3, CD40, FCGR2A, IPTKC, and HLA class II have been ascertained in diverse populations. Also, it is known that the increased risk for the development of coronary aneurysms in populations with European ethnicity is correlated with the genetic variation in the TGF pathway (TGFβ2, TGFβR2, SMAD3) [17].
Heart damage is visible in a significant number of untreated KD patients which can lead to the development of myocardial infarction, unexpected death, or ischemic heart disorder from ectasia (Coronary artery aneurysms) [18, 19]. Early diagnosis is crucial for effective treatment which will result in abolition of the inflammatory process and reduces the risk of coronary artery aneurysms (CAA) rates to approximately 5-10% [20]. CAA is the most important complication of KD patients. In untreated KD children, the disease-associated inflammation modifies the arterial wall which leads to CAA in 25% of them [21]. In developed countries, KD has been reported as the most prevalent cause of acquired heart disease in children [22]. KD shares certain features with other childhood febrile conditions, including, infectious (e.g. staphylococcal and streptococcal toxic shock syndromes, measles and other viral illnesses) and inflammatory conditions which makes differential diagnosis difficult [20]. Although nowadays there are guidelines to assist diagnosis based on clinical signs and symptoms, such as echocardiography, and laboratory variables, distinguishing KD in early stages from other mimicking conditions for treatment and impediment of CAA development remains a momentous mission [23].
Recently, analysis algorithms for differential co-expression network advanced and applied to study the expression data of genes and microRNAs [24, 25]. There is a new functional strategy in systems biology, the weighted gene co-expression network analysis (WGCNA) algorithm, which identifies the most important genes in co-expressed genes in association with a sample trait [26, 27]. Based on similarities in expression profiles of samples, WGCNA makes modules, sub-network regions, and detects those of which contain associated genes [28, 29]. Through analysis of these modules in two different conditions, acute against convalescent, we aimed to identify genes in relation to the acute state that may be used as new therapeutic targets for KD.
2.1. Data selection, preprocessing and detection differentially expressed genes (DEGs) between acute and convalescent samples
In the present study, one expression array, GSE63881, was obtained from NCBI Gene Expression Omnibus (GEO). GSE63881 consists of 341 samples, 171 out of which are acute, and 170 are convalescent. This dataset was produced using Illumina HumanHT-12 V4.0 expression beadchip. Normalizing DFGs of this dataset was performed by quantile normalization method in limma package. For more qualified results, we included only single measurement of each gene, with the aggregate function in the S4Vectors package, which gives an average measurement for each gene’s probes. Using |log2FC| > 0.5 and P.value < 0.05 as the threshold, all the differentially expressed genes were screened out by limma package.
2.2. Evaluating the comparability and detection of outlier samples in WGCNA
Evaluating the comparability of acute and convalescent samples was conducted through softConnectivity function in WGCNA package. SoftConnectivity measures two factors, 1) correlation of expression level of each gene between two datasets, 2) correlation of connectivity of each gene between two datasets. The datasets are comparable if two mentioned correlations are positive and have significant P.value.
To remove outlier samples standardized connectivity (Z. K) method was used and samples which had Z. K score< −2 were excluded from the rest of the analysis.
2.3. Network construction and module detection
According to acute and convalescent samples, two weighted gene co-expression networks was constructed. Since applying pickSoftThreshold function assists in choosing proper soft-threshold power, we have selected soft thresholding power of 6 for providing scale-free topology fit index that reaches values above 0.9. Following calculation of adjacencies, the adjacency results were transformed to Topological Overlap Matrix (TOM) to minimize the effect of noise and spurious associations. TOM of different datasets may have different statistical properties which can affect the results of comparisons. Here scaling of Topological Overlap Matrices was used to mitigate the effect of different statistical properties. Quantile-quantile plot of the TOMs in acute and convalescent datasets was used to discern what scaling is achieved. The results of TOM, as input, and the cutreeDynamic function were utilized in producing dendrogram of genes, and branch cutting, respectively. Minimum module size of 30, and the module detection sensitivity deep Split 2 in blockwiseConsensusModules function used for network construction. Finally, the network modules of acute and convalescent datasets were detected by considering acute network modules as the reference. The function module preservation of WGCNA package was taken advantage to study the preservation of acute modules in convalescents. Zsummary was considered the output of module preservation function for detecting modules that have been preserved in both studies. Modules with Z score more than 10 are well preserved between acute and convalescent samples and accordingly, they were removed from the rest of analysis while the modules with the values of less than 10 are postulated less preserved and considered the result of analysis.
2.4. Functional annotation of tan module
To facilitate the interpretation of their biological mechanisms we carried out functional enrichment analysis for genes of detected module. GO analysis, a regular method in the annotation of large-scale functional enrichment studies, is normally classified into molecular function (MF), biological process (BP), and cellular component (CC) categories. KEGG database was used for functional and biological interpretation of detected genes, and to identify important signaling pathways with P.value < 0.05 and combined score > 10. The PPI network of detected genes was constructed by Search Tool for the Retrieval of Interacting Gene (STRING10.5; https://string-db.org/) with a combined score >0.4, as the cut-off point. Within detected module, those genes with the highest module membership scores have been considered the hub genes of that module, these genes were imported into the Cytoscape [30] to visualize the networks and relationships between genes in the specific cluster.
3.1. Differentially expressed genes (DEGs) screening
After preprocessing and quality assessment, using limma package DEGs with cutoff |log2FC| > 0.5 and P.value < 0.05 as the threshold we detected 374 upregulated and 687 downregulated genes. The DEGs of this dataset was shown as volcano plot in Figure 1.
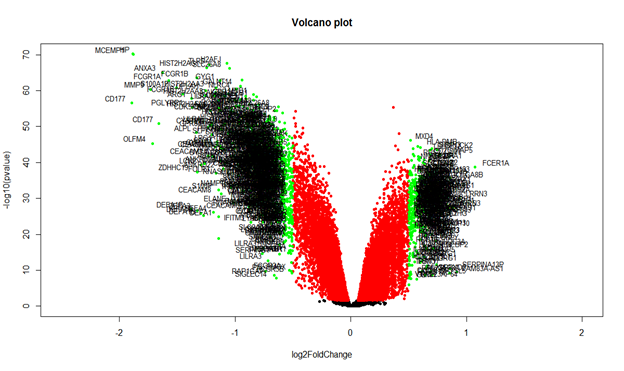
Figure 1: The volcano plot of differentially expressed genes of GSE63881 between acute and convalescent samples.
3.2. WGCNA prerequisites, outlier samples, batch effects, comparability
One of the key points in using the WGCNA package is the number of samples, the number of samples for construction of weighted gene co-expression network should be at least 15 while the number of samples included in current study was 171 and 170 for acute and convalescent status, respectively. Since only one dataset with one platform and one laboratory condition was used, the batch effect was not observed. Fulfilling the WGCNA package requirements, two and five outlier samples, based on the Z.K score, were excluded from the acute and convalescent datasets respectively (Figure 2). Furthermore, datasets comparability was evaluated by the softConnectivity function which provides positive correlation values when there is high comparability between two datasets. Our datasets were comparable as the overall gene expression correlation (cor=0.93, P.value<1e−200), and the overall gene connectivity (cor=0.7, P.value<1e−200) were significant.( Figure 3).
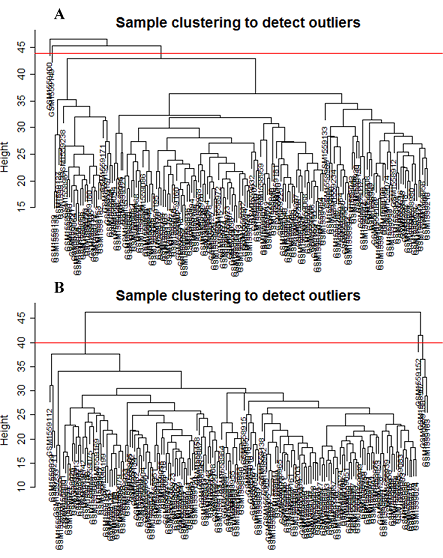
Figure 2: Clustering dendrogram of samples based on their euclidean distance and detecting outlier samples. (A) Acute samples, (B) Convalescent samples
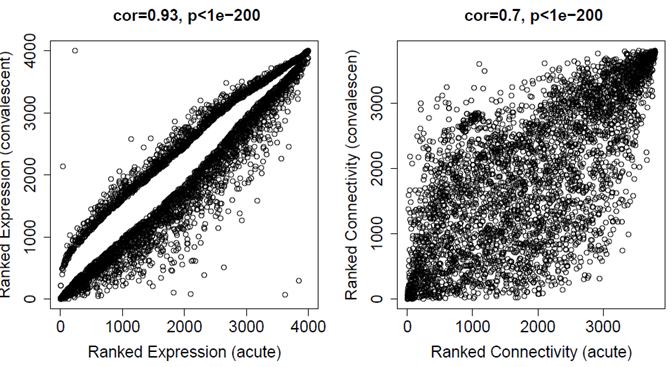
Figure 3: Results of softConnectivity. Correlations are positive and the p-values are significant for both datasets. In addition correlations and p-values are better for expression than for connectivity.
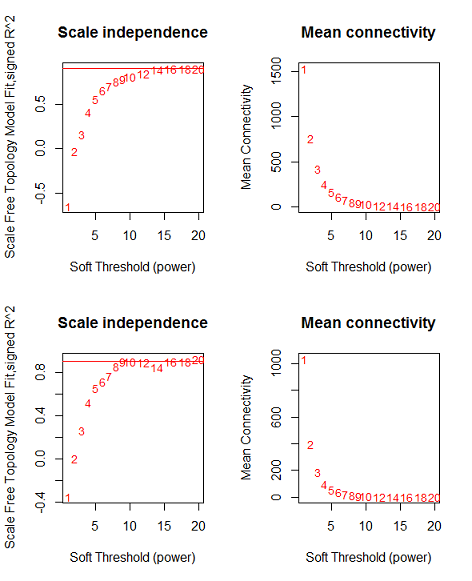
Figure 4: network topology for various soft-thresholding powers, the left panel shows the scale-free fit index and the right panel displays the mean connectivity for (A) acute dataset, (B) Convalescent dataset.
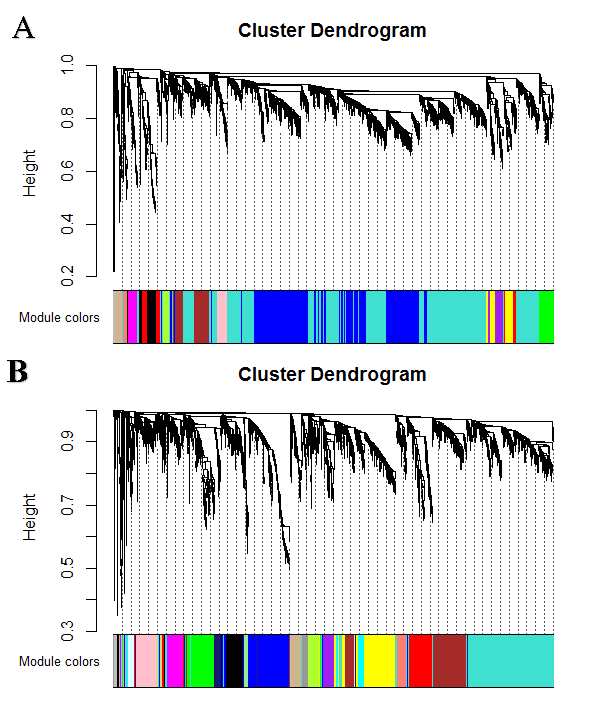
Figure 5: Clustering dendrogram of genes, with dissimilarity based on topological overlap, together with assigned module colors for (A) acute dataset, (B) convalescent dataset
3.3. Identification of modules able to distinguish acute from convalescent datasets
Acute and convalescent datasets exhibited a scale-free topology with the powers equal to 6 for both acute and convalescent samples and the Scale-free Topology Fit Index reached values above 0.9 for low powers (< 30). These results also show that the batch-effects were not present in our datasets (Figure 4). After constructing weighted gene co-expression networks for each acute and convalescent expression dataset, the modules of these datasets were identified. Based on the WGCNA package, a module is a group of strongly co-expressed genes, which have similar biochemical and functional properties or belong to similar pathways. By hierarchical clustering, 14 modules were identified for the acute dataset as the reference network and also 15 modules for convalescent samples as the test network, All of which had different number of genes which are labelled by different colors and are shown in Figure 5. By considering convalescent dataset a test network, we evaluated the preservation level across the two networks by finding preserved and non-preserved modules in two datasets, . As it was expected, except for one module, they were well preserved between two mentioned expression datasets. Therefore, this module was potentially related to the KD disease pathogenesis. Qualifying the module preservation level was conducted through taking advantage of modulePreservation function from WGCNA package. For each module, a Z-score representing the preservation level was calculated. Modules with Z-score higher than 10 were considered well preserved between two datasets, whereas modules with Z-scores <10 were assumed less preserved and distinguishing acute from the convalescent condition. (Figure 6)
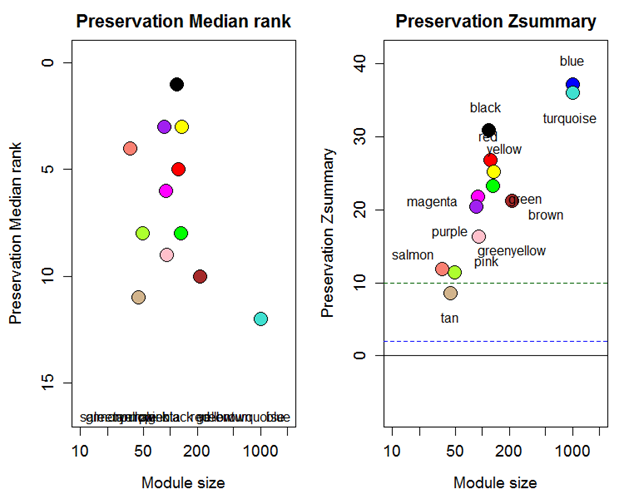
Figure 6: The medianRank and Zsummary statistics of module preservation of acute modules in convalescent modules (y-axis) vs. module size (x-axis). In the left panel Z score for tan module is lower than 10 and considered as not preserved module in transition from acute to convalescent phase of KD.
3.4. Identification of hub genes and functional analysis
Since we found tan module non-preserved between acute and convalescent datasets, it seemed that tan module-related genes play an important role in the acute phase of disease. We set out to identify genes related to this module (Table 1). According to the results of gene ontology, the most important signaling pathways pertained to the innate immune defense against the infectious agent, and in particular, in the process that neutrophils, as professional killers of invading pathogens to the human organism act by releasing various antimicrobial proteins during degranulation (Figure 7A). The cenplot that shows the relation between signaling pathways and tan module genes is shown in Figure 7B. The PPI network of detected genes was constituted by STRING (Figure 8A), 36 nodes and 102-edged related hub genes in PPI were recognized. After PPI network establishment, the PPI data imported into Cytoscape software and the interactions of hub genes were detected. (Figure 8B). The genes including, BPI, CAMP, DEFA1, DEFA3, DEFA4, ELANE, LCN2, MMP8, and ORM2 were the most interacting genes identified as hub genes.
Table 1: Genes identified in transition from acute to convalescent phase.
|
Module |
Hub Genes |
|
Tan |
CAMP, AZU1, OLR1, RNASE3, CEACAM6, SLC22A16, CEACAM8, BEX1, DEFA4, MS4A3, DEFA1B, DEFA1, ELANE, DEFA3, ABCA13, LCN2, CTSG, TCN1, BPI, PRTN3, CEBPE, LTF, COL17A1, TACSTD2, PTPN20, MPO, HYAL3, ATP8B4, TFF3, OLFM4, SERPINB10, MMP8, TCTEX1D1, ORM2, CRISP3, ORM1 |
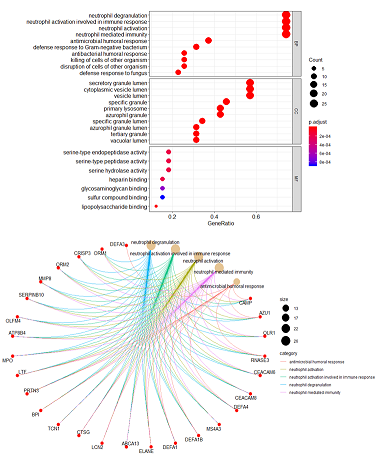
Figure 7: Functional enrichment analysis of tan module genes: (A) Gene ontology analysis of all genes in tan module. (B) cnetplot of all genes in tan module that depicts the linkages of genes and most important signaling pathways.
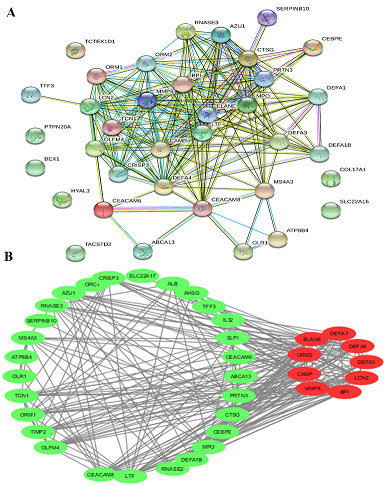
Figure 8: The PPI network of detected genes that was analyzed by String software. 36 nodes and 102 edged in the PPI network. BPI, CAMP, DEFA1, DEFA3, DEFA4, ELANE, LCN2, MMP8 and ORM2 are the most interacted genes and identified as hub genes.
To analyze complicated diseases with obscure etiology such as KD, a comprehensive study is required to cover all aspects of the disease, including molecular phenomena, pathogenicity, physiological side effects, and cellular alterations. Since there is no complete data about KD, molecular analysis alongside clinical studies can help us explain etiology of diseases and achieve new biomarkers and safe therapy [31, 32]. In this study, we employed WGCNA package on the expression data obtained from GSE63881 of the acute and convalescent phase of KD patients. By WGCNA package, we discovered that one out of 14 network modules (Tan module) in the acute network were not preserved in the convalescent KD network. This gene module may play a significant part in KD pathogenesis and consequently, it is probable for the hub gens within this module to serve as new diagnostic or therapeutic purposes. Afterward, we summarized the list of genes within this module through distinguishing only the hub genes or the most KD-associated genes according to the WGCNA package. Functional enrichment analysis of genes in Tan module revealed that the most significant GO terms are related to the innate immune response against infectious agents.
Although the etiology of KD remains unknown so far, several lines of clinical and epidemiological data suggest an environmental trigger such as epidemic occurrence, change in season, and temporal clustering of cases [33, 34]. The new hypothesis of the etiology of KD explains that an abnormal response to undisclosed infectious diseases in genetically susceptible children may be the trigger of the disease. One of the first hypotheses based on the implication of an infectious agent in KD was superantigen-mediated etiology that came from the similarities in clinical features and immunological reactions between KD, toxic shock syndrome (TSS), and streptococcal toxic shock syndrome (STSS), but further studies have failed to confirm these findings [35-38]. The scientific society have so far failed to achieve an understanding about the infectious origin of KD (viral, bacterial or fungal), or identification of the underlying immune mechanisms behind KD, accordingly, other possible environmental triggers have been assessed by various research groups to determine the role of the resident gut microbiota as a potential contributor to KD. Many pieces of research and microbiologic observations fortified this hypothesis that heterogeneity and abnormalities in the gut microbiota composition may set off or contribute to the development of KD [39].
We detected alpha-defensins in our detected module. Based on this result, we suggest that an unknown infectious agent or an unknown intestinal microbiota, may induce KD. Alpha-defensins, host defence peptides that supply protection against viral, bacterial, and fungal infections, are mostly found in neutrophils and Paneth cells of the small intestine. If host primary defense mechanisms are incapable to identify and eliminate the infectious agent, chemokines and other warning signals summon circulating polymorphonuclear leukocytes (PMNs) to the Infection environment. In humans, antimicrobial peptides that belong to the alpha-defensin (DEFA1-4) and cathelicidin (hCAP-18/LL-37 encoded by the CAMP gene) families are the main components of the PMN apparatus. The α-defensins and hCAP-18/LL-37 are stored in the primary (azurophil) granules and secondary (specific) granules, respectively. This different storage sites is an indication for their respective scene of action because azurophil granules are preferentially derived from phagosome and specific granules are mainly in the extracellular environment [40]. The antimicrobial function of alpha-defensins during the viral, bacterial, protozoa (Entamoeba histolytica), and fungal infections in small intestine is well explained [39, 41-43]. Bactericidal/permeability-increasing protein (BPI), neutrophil elastase (ELANE), proteinase 3 (PRTN3), Neutrophil Gelatinase-Associated Lipocalin encoded by the LCN2 gene, and cathepsin G (CTSG) are other components of azurophil granule that participate in the killing pathogens in phagosomes [40, 44, 45]. It has been shown that systemic upregulation of LCN-2 associate with the development of several inflammatory disease, such as peritonitis, atherosclerosis, and vasculitis/Kawasaki disease [46]. With regard to the NOD-like receptor signaling pathway in KEGG results, it is recognized that NOD2 (a pathogen recognition receptor of the NOD-like receptor family) controls the expression of a subset of paneth cell-expressed alpha-defensins, and the cumulation of both microbiota and pathogenic infection in the terminal ileum in mice [47].
In the present study, we attempted to figure out potential molecular mechanisms in KD by using a comprehensive algorithm for differential co-expression network. The non-preserved module, tan module, has possibly been associated with the pathogenesis of KD. Functional enrichment analyses demonstrated that the innate immune defense against the infectious agent, and, the processes that neutrophils kill invading pathogens by mediating antimicrobial humoral response, involved in the pathogenesis of KD. Additionally, hub genes may be used as transcriptional biomarkers or potential therapeutic targets.
Burns, J.C. and M.P. Glodé, Kawasaki syndrome. The Lancet, 2004. 364(9433): p. 533-544. 16814-1
View ArticleKuo, H.C., Preventing coronary artery lesions in Kawasaki disease. Biomed J, 2017. 40(3): p. 141-146. PMid:28651735
View Article PubMed/NCBIMori, M., et al., Two-generation Kawasaki disease: mother and daughter. The Journal of pediatrics, 2001. 139(5): p. 754-756. PMid:11713463
View Article PubMed/NCBINewburger, J.W., et al., Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Circulation, 2004. 110(17): p. 2747-2771. PMid:15505111
View Article PubMed/NCBIKawasaki, T., Acute febrile mucocutaneous syndrome with lymphoid involvement with specific desquamation of the fingers and toes in children. Arerugi, 1967. 16: p. 178-222.
Kato, H., et al., Long-term consequences of Kawasaki disease: a 10-to 21-year follow-up study of 594 patients. Circulation, 1996. 94(6): p. 1379-1385. PMid:8822996
View Article PubMed/NCBIMakino, N., et al., Descriptive epidemiology of Kawasaki disease in Japan, 2011-2012: from the results of the 22nd nationwide survey. J Epidemiol, 2015. 25(3): p. 239-45. PMid:25716368
View Article PubMed/NCBILue, H.C., et al., Estimation of the incidence of Kawasaki disease in Taiwan. A comparison of two data sources: nationwide hospital survey and national health insurance claims. Pediatr Neonatol, 2014. 55(2): p. 97-100. PMid:23890670
View Article PubMed/NCBIDu, Z.D., et al., Epidemiologic study on Kawasaki disease in Beijing from 2000 through 2004. Pediatr Infect Dis J, 2007. 26(5): p. 449-51. PMid:17468660
View Article PubMed/NCBIKim, G.B., et al., Epidemiology and Clinical Features of Kawasaki Disease in South Korea, 2012-2014. Pediatr Infect Dis J, 2017. 36(5): p. 482-485. PMid:27997519
View Article PubMed/NCBIHarnden, A., et al., Kawasaki disease in England: ethnicity, deprivation, and respiratory pathogens. Pediatr Infect Dis J, 2009. 28(1): p. 21-4. PMid:19145710
View Article PubMed/NCBIHolman, R.C., et al., Kawasaki syndrome hospitalizations among children in Hawaii and Connecticut. Archives of pediatrics & adolescent medicine, 2000. 154(8): p. 804-808. PMid:10922277
View Article PubMed/NCBIShulman, S.T. and A.H. Rowley, Kawasaki disease: insights into pathogenesis and approaches to treatment. Nat Rev Rheumatol, 2015. 11(8): p. 475-82. PMid:25907703
View Article PubMed/NCBIRamphul, K. and S.G. Mejias, Kawasaki disease: a comprehensive review. Archives of medical sciences. Atherosclerotic diseases, 2018. 3: p. e41-e45. PMid:30775588
View Article PubMed/NCBINakamura, Y., et al., Epidemiologic features of Kawasaki disease in Japan: results of the 2009-2010 nationwide survey. J Epidemiol, 2012. 22(3): p. 216-21. PMid:22447211
View Article PubMed/NCBIDergun, M., et al., Familial occurrence of Kawasaki syndrome in North America. Arch Pediatr Adolesc Med, 2005. 159(9): p. 876-81. PMid:16143748
View Article PubMed/NCBIHedrich, C.M., A. Schnabel, and T. Hospach, Kawasaki Disease. Frontiers in pediatrics, 2018. 6: p. 198-198. PMid:30042935
View Article PubMed/NCBIBaer, A.Z., et al., Prevalence of coronary artery lesions on the initial echocardiogram in Kawasaki syndrome. Archives of pediatrics & adolescent medicine, 2006. 160(7): p. 686-690. PMid:16818833
View Article PubMed/NCBIDajani, A.S., et al., Diagnosis and therapy of Kawasaki disease in children. Circulation, 1993. 87(5): p. 1776-1780. PMid:8491037
View Article PubMed/NCBIWright, V.J., et al., Diagnosis of Kawasaki Disease Using a Minimal Whole-Blood Gene Expression Signature. JAMA Pediatr, 2018. 172(10): p. e182293. PMid:30083721
View Article PubMed/NCBIKato, H., et al., Long-term consequences of Kawasaki disease. A 10- to 21-year follow-up study of 594 patients. Circulation, 1996. 94(6): p. 1379-85. PMid:8822996
View Article PubMed/NCBITaubert, K.A., A.H. Rowley, and S.T. Shulman, Nationwide survey of Kawasaki disease and acute rheumatic fever. J Pediatr, 1991. 119(2): p. 279-82. 80742-5
View ArticleMcCrindle, B.W., et al., Diagnosis, Treatment, and Long-Term Management of Kawasaki Disease: A Scientific Statement for Health Professionals From the American Heart Association. Circulation, 2017. 135(17): p. e927-e999.
Wang, Y.R. and H. Huang, Review on statistical methods for gene network reconstruction using expression data. Journal of theoretical biology, 2014. 362: p. 53-61. PMid:24726980
View Article PubMed/NCBILin, C.-C., et al., A cross-cancer differential co-expression network reveals microRNA-regulated oncogenic functional modules. Molecular biosystems, 2015. 11(12): p. 3244-3252. PMid:26448606
View Article PubMed/NCBIGiulietti, M., et al., Identification of candidate miRNA biomarkers for pancreatic ductal adenocarcinoma by weighted gene co-expression network analysis. Cellular Oncology, 2017. 40(2): p. 181-192. PMid:28205147
View Article PubMed/NCBIZhang, B. and S. Horvath, A general framework for weighted gene co-expression network analysis. Statistical applications in genetics and molecular biology, 2005. 4(1). PMid:16646834
View Article PubMed/NCBIGargalovic, P.S., et al., Identification of inflammatory gene modules based on variations of human endothelial cell responses to oxidized lipids. Proceedings of the National Academy of Sciences, 2006. 103(34): p. 12741-12746. PMid:16912112
View Article PubMed/NCBICarlson, M.R., et al., Gene connectivity, function, and sequence conservation: predictions from modular yeast co-expression networks. BMC genomics, 2006. 7(1): p. 40. PMid:16515682
View Article PubMed/NCBISmoot, M.E., et al., Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics, 2010. 27(3): p. 431-432. PMid:21149340
View Article PubMed/NCBIKato, H., et al., Adult coronary artery disease probably due to childhood Kawasaki disease. The Lancet, 1992. 340(8828): p. 1127-1129. 93152-D
View ArticleRowley, A.H., Kawasaki disease: novel insights into etiology and genetic susceptibility. Annual review of medicine, 2011. 62: p. 69-77. PMid:20690826
View Article PubMed/NCBIBurns, J.C., et al., Seasonality and temporal clustering of Kawasaki syndrome. Epidemiology, 2005. 16(2): p. 220-5. PMid:15703537
View Article PubMed/NCBIMenikou, S., P.R. Langford, and M. Levin, Kawasaki Disease: The Role of Immune Complexes Revisited. 2019. 10(1156). PMid:31263461
View Article PubMed/NCBIMatsubara, K. and T. Fukaya, The role of superantigens of group A Streptococcus and Staphylococcus aureus in Kawasaki disease. Curr Opin Infect Dis, 2007. 20(3): p. 298-303. PMid:17471041
View Article PubMed/NCBICurtis, N., B. Chan, and M. Levin, Toxic shock syndrome toxin-secreting Staphylococcus aureus in Kawasaki syndrome. Lancet, 1994. 343(8892): p. 299. 91149-5
View ArticleMorita, A., et al., Serologic Evidence That Streptococcal Superantigens Are Not Involved in the Pathogenesis of Kawasaki Disease. 1997. 41(11): p. 895-900. PMid:9444333
View Article PubMed/NCBIGupta-Malhotra, M., et al., Antibodies to highly conserved peptide sequence of staphylococcal and streptococcal superantigens in Kawasaki disease. Experimental and Molecular Pathology, 2004. 76(2): p. 117-121. PMid:15010289
View Article PubMed/NCBIEsposito, S., I. Polinori, and D. Rigante, The Gut Microbiota-Host Partnership as a Potential Driver of Kawasaki Syndrome. Front Pediatr, 2019. 7: p. 124. PMid:31024869
View Article PubMed/NCBILehrer, R.I., Primate defensins. Nat Rev Microbiol, 2004. 2(9): p. 727-38. PMid:15372083
View Article PubMed/NCBIHolly, M.K. and J.G. Smith, Paneth Cells during Viral Infection and Pathogenesis. Viruses, 2018. 10(5). PMid:29701691
View Article PubMed/NCBIGacser, A., et al., Induction of human defensins by intestinal Caco-2 cells after interactions with opportunistic Candida species. Microbes Infect, 2014. 16(1): p. 80-5. PMid:24095867
View Article PubMed/NCBICobo, E.R., et al., Entamoeba histolytica Alters Ileal Paneth Cell Functions in Intact and Muc2 Mucin Deficiency. Infect Immun, 2018. 86(7). PMid:29685982
View Article PubMed/NCBIEick, S., et al., Lack of cathelicidin processing in Papillon-Lefevre syndrome patients reveals essential role of LL-37 in periodontal homeostasis. Orphanet J Rare Dis, 2014. 9: p. 148. PMid:25260376
View Article PubMed/NCBILe Cabec, V., et al., Targeting of proteins to granule subsets is determined by timing and not by sorting: The specific granule protein NGAL is localized to azurophil granules when expressed in HL-60 cells. Proc Natl Acad Sci U S A, 1996. 93(13): p. 6454-7. PMid:8692836
View Article PubMed/NCBIBiezeveld, M.H., et al., Sustained activation of neutrophils in the course of Kawasaki disease: an association with matrix metalloproteinases. Clin Exp Immunol, 2005. 141(1): p. 183-8. PMid:15958085
View Article PubMed/NCBICaruso, R., et al., NOD1 and NOD2: signaling, host defense, and inflammatory disease. Immunity, 2014. 41(6): p. 898-908. PMid:25526305
View Article PubMed/NCBI