S.Janani
Email: s.jananiphysics@gmail.com
© 2019 Sift Desk Journals. All Rights Reserved
VOLUME: 4 ISSUE: 4
Page No: 463-477
S.Janani
Email: s.jananiphysics@gmail.com
N.Sudhaa , S.Janania*, G.K.Vanathi Nachiyara, T.Kavithab
a Department of Physics, Sri Sarada College for Women (Autonomous), Salem-16, Tamil Nadu, India.
b Department of Physics, Government Arts College for Women, Salem-636008, Tamil Nadu, India.
S.Janani, DENSITY FUNCTIONAL THEORY ANALYSIS AND MOLECULAR DOCKING EVALUATION OF 8-CHLOROQUINOLINE 2-CARBALDEHYDE (2020)Journal of Computational Chemistry & Molecular Modeling 4(4)
DFT/B3LYP method 6-31G (d,p) basis set. The geometrical parameters have been obtained using same basis set. The interpretations of vibrational assignments have been calculated by VEDA program. The 1H and 13C NMR chemical shifts are calculated by GIAO method. The vibrational optical polarization characteristics were studied by VCD spectrum. The electronic and charge transfer properties have been explained on the basis of highest occupied molecular orbitals (HOMOs), lowest unoccupied molecular orbitals (LUMOs) and UV absorption spectrum. Natural Bond Orbital (NBO) analysis has been performed for analyzing charge delocalization. In addition, Mulliken charges and MEP are performed in the molecule to identify the reactive sites. The values of dipole moment, polarizability and hyperpolarizability have been used to calculate nonlinear activity. The molecular docking studies were performed for the title molecule with different proteins by using AutoDock software.
Keywords: DFT, Vibrational assignments, MEP, HOMO-LUMO, NBO, Molecular docking.
Quinoline derivatives have wide applications such as live saving drugs, optical switches in nonlinear optics, sensors in electrochemistry and in the field of inorganic chemistry [1, 2]. Nitrogen and oxygen containing heterocyclic compounds have received considerable attention due to their wide range of pharmacological activity [3]. Quinoline is a heterocyclic aromatic organic compound. It has several anti-malarial derivatives, like quiline, chloroquine, amodiaquine, and primaquine [4]. The current interest in the development of new antimicrobial agents can be partially ascribed both to the increasing emergence of bacterial resistance to antibiotic therapy and to newly emerging pathogens [5, 6]. In synthetic medicinal chemistry the quinoline motif is widely exploited revealing a spectrum of activity covering antimalarial [7], anticancer [8], antifungal, antibacterial, antiprotozoic, antibiotic [9] and anti-HIV [10] effects. Quinoline compounds are known to be effective antimicrobial compounds [11]. 8-chlorquinoline 2-carbaldehyde is an organic compound with formula C10H6ClNO and molecular weight is 191.614 g/mol. These aldehydes have wide range of ligand-protein activity. 8-Chloroquinoline 2- Carbaldehyde a derivative of quinoline [12] is treated for both Parkinson’s disease [13] and schizophrenia [14].
Parkinson’s disease is the second most common neurodegenerative disorder and the most common movement disorder [15]. Characteristics of Parkinson’s disease are progressive loss of muscle control, which leads to trembling of the limbs and head while at rest, stiffness, slowness and impaired balance. As symptoms it may become difficult to walk, talk and complete simple tasks [16].
Schizophrenia is a mental disorder characterized by abnormal behavior, strange speech and a decreased ability to understand reality [17]. Other symptoms include false belief, unclear or confused thinking and reduced social engagement. People with schizophrenia often have additional mental health problems such as anxiety and depression [18]. The 8CQ2C is docked with two different proteins like 2V60 and 4MRW.
The title compound is optimized by DFT/B3LYP method with 6-31 G(d,p) basis set. The molecular structural parameters like bond length and bond angle, vibrational assignments, HOMO - LUMO and UV analysis are studied. 1H-NMR, 13C-NMR and VCD spectra are studied. Also, Mulliken, MEP and NBO calculations are studied to identify the reactive sites. In addition hyperpolarizability calculations are found to validate for NLO activity. Molecular docking investigation has been performed to find out the hydrogen bond lengths, binding energy and drug activity of the molecule.
The computational calculations are carried out by DFT method using Gaussian 09 [19] program and Gauss view visualization software [20]. 8CQ2C molecule has been completely optimized by the DFT/B3LYP method with 6-31 G (d,p) basis set. The vibrational assignments were obtained by using Veda4 software program [21]. The electronic properties such as UV absorption, HOMO (Highest Occupied Molecular Orbital) and LUMO (Lowest Unoccupied Molecular Orbital) energies were calculated using DFT method. In addition, Mulliken charges, the dipole moment and nonlinear optical (NLO) activity, such as the first hyperpolarizability, MEP analysis of the title molecule are computed. The NMR chemical shift H and C were carried out using GIAO method in the combination of DFT/B3LYP method with 6-31 G (d, p) basis set. The molecular docking were obtained by the Autodock Tools version 1.5.6 software package [22] and the docking results are viewed and analyzed using pymol [23], and Discovery studio [24] visualization software.
3.1. Molecular geometry
The bond parameters (bond length and bond angles) of the 8CQ2C molecules are listed in Table 1 using DFT/B3LYP method with 6-31 G (d,p) basis set. The optimized molecular structure was obtained from Gaussian 09 and viewed by Gauss View 5.0 programs. The optimized structure is shown in Fig 1. The global minimum energy E= - 974.8020 Hartrees. The molecular structure of 8CQ2C belongs to C1 point group symmetry. The title compound having nine C-C, six C-H, one C=O, two C-N and one C-Cl bond lengths and total number of atoms 19. All the bond lengths and angles are in the normal ranges [25].
Table 1. Geometrical parameters from DFT/B3LYP method.
|
Bond Length |
Theoretical bond Length (Å) |
Bond Angle |
Theoretical bond angle (deg) |
|
C1-C2 |
1.372 |
C2-C1-C6 |
120.376 |
|
C1-C6 |
1.416 |
C2-C1-H7 |
120.647 |
|
C1-H7 |
1.100 |
C6-C1-H7 |
118.97 |
|
C2-C3 |
1.422 |
C1-C2-C3 |
120.509 |
|
C2-Cl15 |
1.760 |
C1-C2-Cl15 |
120.883 |
|
C3-C4 |
1.418 |
C3-C2-Cl15 |
118.606 |
|
C3-N16 |
1.422 |
C2-C3-C4 |
119.095 |
|
C4-C5 |
1.421 |
C2-C3-N16 |
121.700 |
|
C4-C8 |
1.422 |
C4-C3-N16 |
119.203 |
|
C5-C6 |
1.373 |
C3-C4-C5 |
119.210 |
|
C5-H9 |
1.100 |
C3-C4-C8 |
119.059 |
|
C6-H10 |
1.100 |
C5-C4-C8 |
121.730 |
|
C8-C11 |
1.372 |
C4-C5-C6 |
120.455 |
|
C8-H13 |
1.100 |
C4-C5-H9 |
118.644 |
|
C11-C12 |
1.416 |
C6-C5-H9 |
120.899 |
|
C11-H14 |
1.100 |
C1-C6-C5 |
120.351 |
|
C12-N16 |
1.372 |
C1-C6-H10 |
119.002 |
|
C12-C17 |
1.540 |
C5-C6-H10 |
120.645 |
|
C17-H18 |
1.070 |
C4-C8-C11 |
120.549 |
|
C17-O19 |
1.430 |
C4-C8-H13 |
118.591 |
|
|
|
C11-C8-H13 |
120.859 |
|
|
|
C8-C11-C12 |
120.366 |
|
|
|
C8-C11-H14 |
120.649 |
|
|
|
C12-C11-H14 |
118.983 |
|
|
|
C11-C12-N16 |
120.323 |
|
|
|
C11-C12-C17 |
118.991 |
|
|
|
N16-C12-C17 |
120.685 |
|
|
|
C3-N16-C12 |
120.496 |
|
|
|
C12-C17-H18 |
109.471 |
|
|
|
C12-C17-O19 |
109.471 |
|
|
|
H18-C17-O19 |
109.471 |
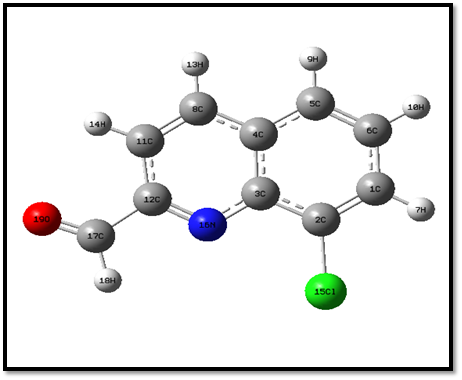
Fig 1. Optimized structure of 8CQ2C
The general bond lengths of C=C are obtained from the range 1.371 to 1.394 Å [26]. The aromatic ring C=C bond length C1-C2, C5-C6, C8=C11 falls on the range 1.372 Å which agree with general range. The general values of C-C bond lengths are obtained from 1.339 –1.417 Å [27]. The bond lengths of C1-C6, C3-C4, C2-C3, C4-C5, C4-C8 = 1.421 Å are due to single bond. The general value of C-N bond length is obtained from 1.325 to 1.437 Å. The bond length C12=N16 is obtained from 1.422 Å is due to the attached electronegative atom. The maximum bond length is C2-Cl15=1.76 Å which it is attached to electronegative atom. The general value of C-H fall in the range 0.93 Å. In the present case bond lengths C1-H7, C6-H10 and C17-H18 are increased from the range 1.1 Å. The increasing band length C17-O19 =1.43 Å is attached with double bond of electronegative atom.
The calculated approximate bond angles of C5-C4-C8, C2-C3-N16 are exactly 121° in the benzene ring. The bond angles C2-C1-C6, C2-C1-C3, C2-C1-H7, C1-C2-Cl15, C4-C5-C6, C6-C5-H9, C1-C6-C5, C5-C6-H10, C4-C8-C11, C8-C11-H14, N16-C12-N16 and C3-N16-C12 all are same values for 120°respectively. Hence, the homonuclear bond lengths (C1-C2, C5-C6, C1-C6, C3-C4, C2-C3, C4-C5, C4-C8 and C11-C12) are higher than the heteronuclear bond lengths (C12-N12, C1-H7, C6-H10 and C17-H18). The reason is same charges are repulsive and opposite charges are attractive.
3.2. Vibrational Assignments
8CQ2C consists of total number of 19 atoms are existing 51 normal modes of vibrations. These vibrational frequencies are computed by DFT/B3LYP method 6-31G (d, p) basis set. The calculated vibrational frequencies (Unscaled and Scaled), IR intensity, Raman activity are given in Table 2. The theoretical spectrum of FT-IR and FT-Raman are shown in Fig 2.
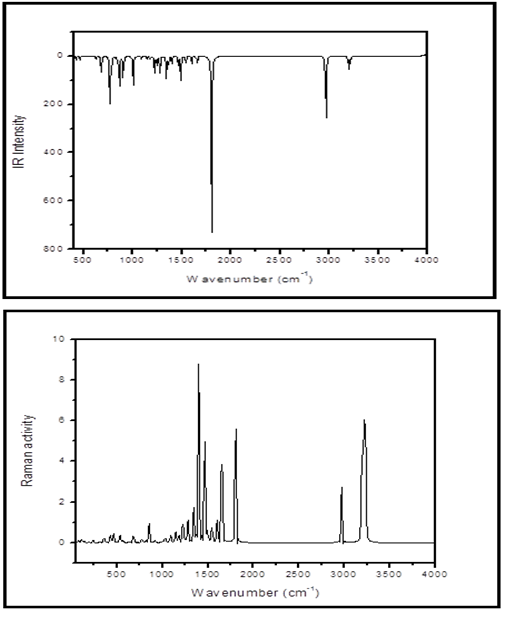
Fig 2. Theoretical FT-IR and FT-Raman spectra of 8CQ2C
Table 2. Theoretical vibrational assignments of 8CQ2C with DFT/B3LYP method 6-31G (d, p) basis set.
|
Sl.No |
Calculated frequencies (cm-1) |
IR intensity |
Raman activity |
% Potential Energy Distribution (PED) |
|
|
Unscaled frequency |
Scaled frequency |
||||
|
1 |
3230 |
3088 |
1.9915 |
122.5108 |
νCH(100) |
|
2 |
3223 |
3081 |
3.8047 |
190.6865 |
νCH(96) |
|
3 |
3206 |
3065 |
15.0989 |
168.0079 |
νCH(93) |
|
4 |
3190 |
3050 |
7.1215 |
133.8051 |
νCH(87) |
|
5 |
3188 |
3048 |
1.8775 |
12.2100 |
νCH(87) |
|
6 |
2967 |
2836 |
81.6869 |
113.9178 |
νCH(100) |
|
7 |
1804 |
1723 |
254.0920 |
187.5745 |
νOC(91) |
|
8 |
1658 |
1585 |
7.8472 |
58.4751 |
νCC(42) |
|
9 |
1648 |
1575 |
2.9679 |
95.9017 |
νCC(55) |
|
10 |
1603 |
1532 |
9.1013 |
29.6118 |
νCC(49) |
|
11 |
1544 |
1476 |
13.1408 |
25.2022 |
νCC(21); βHCC(27) |
|
12 |
1493 |
1427 |
37.6452 |
14.7409 |
νNC(36); βHCC(16) |
|
13 |
1465 |
1401 |
12.7348 |
146.5212 |
βHCC(50) |
|
14 |
1401 |
1339 |
9.2703 |
225.6608 |
νCC(62); βCNC(10) |
|
15 |
1391 |
1330 |
0.2543 |
30.4168 |
βHCO(71) |
|
16 |
1364 |
1304 |
9.9375 |
2.4671 |
νCC(41) |
|
17 |
1343 |
1284 |
27.8245 |
45.3592 |
νNC(16); νCC(17) |
|
18 |
1278 |
1222 |
29.0341 |
33.7513 |
νNC(10); νCC(21); βHCC(13) |
|
19 |
1245 |
1190 |
10.3566 |
4.2636 |
νCC(29); βHCC(40) |
|
20 |
1223 |
1169 |
22.4116 |
25.1403 |
βCNC(10); βHCC(47) |
|
21 |
1181 |
1129 |
4.1477 |
9.6040 |
βHCC(60) |
|
22 |
1145 |
1095 |
4.5390 |
16.3995 |
νCC(10); βHCC(62) |
|
23 |
1090 |
1042 |
4.7019 |
9.6352 |
νCC(59); βHCC(17) |
|
24 |
1032 |
987 |
0.2764 |
6.4745 |
τHCCC(61); τHCCC(11) |
|
25 |
1008 |
964 |
0.2619 |
0.0587 |
τCCCC(77) |
|
26 |
1007 |
963 |
38.7542 |
2.1605 |
νClC(25); βCNC(11); βCCC(31) |
|
27 |
987 |
944 |
0.7726 |
0.3712 |
τHCCC(71); δCCCC(11) |
|
28 |
919 |
879 |
0.4471 |
2.9488 |
τHCCC(84) |
|
29 |
904 |
864 |
27.7668 |
0.2652 |
νCC(23); βCCC(21) |
|
30 |
867 |
829 |
39.8479 |
1.6734 |
τHCCC(77) |
|
31 |
852 |
815 |
11.2384 |
23.6542 |
νCC(12); βCCC(14); βCCN(12) |
|
32 |
819 |
783 |
1.2290 |
2.3140 |
τHCCC(16); τCCCC(44) |
|
33 |
780 |
746 |
26.8303 |
2.7301 |
τHCCC(73); τCCCC(15) |
|
34 |
769 |
735 |
54.1753 |
2.2194 |
νCC(12); βCCN(27) |
|
35 |
682 |
652 |
26.7380 |
8.9252 |
νCCl(14); βCCC(33) |
|
36 |
671 |
641 |
0.2195 |
0.0725 |
δCCCC(40); τCCCN(40); τCCCC(21) |
|
37 |
627 |
599 |
4.3908 |
0.8504 |
νCC(13); βCCO(32); βCCN(13) |
|
38 |
569 |
543 |
0.0049 |
2.4302 |
τHCCC(13); δCCCC(60) |
|
39 |
536 |
512 |
0.2083 |
9.8838 |
βCCC(55) |
|
40 |
514 |
419 |
0.3966 |
0.5207 |
δCCCC(12); τCCCN(23); τCCCC(35) |
|
41 |
463 |
443 |
5.2396 |
9.5045 |
βClC(15); βCCC(20) |
|
42 |
431 |
412 |
0.2816 |
3.3354 |
δCCCC(10); τCNCC(64) |
|
43 |
429 |
410 |
5.8515 |
5.6801 |
βCCl(51); βCCC(42) |
|
44 |
361 |
345 |
1.2694 |
5.1779 |
νCCl(14); βCCC(14); βCCO(16); βCCN(21) |
|
45 |
312 |
298 |
1.5142 |
0.7750 |
τHCCC(11); τCCCN(58) |
|
46 |
239 |
228 |
7.9312 |
2.8326 |
βCCCl(39) |
|
47 |
213 |
204 |
9.5952 |
0.0534 |
δCCCC(16); τCCCN(40) |
|
48 |
192 |
183 |
3.8015 |
0.3850 |
δCCCC(60) |
|
49 |
142 |
136 |
1.8011 |
1.3247 |
βCCCl(64) |
|
50 |
113 |
108 |
0.9963 |
3.3120 |
τCCCC(45); δCCCC(11) |
|
51 |
78 |
75 |
0.7571 |
2.4633 |
τCCCC(11); δCCCC(75) |
C-H Vibrations
Heterocyclic aromatic compound and its derivatives are structurally very close to benzene. The C–H stretching vibrations for hetero aromatic molecule appear in the region from 3100-3000 cm-1 [28, 29]. The C-H vibrations for 8CQ2C were observed at 3088, 3081, 3065, 3050, 3048 cm-1 with %PED is reported. The general value for in plane bending of C–H is the range of 1000–1300 cm−1[30]. In the present compound C-H in plane bending are observed at 1190, 1169, 1129 and 1095 cm-1 with including % PED conforming CH bending.
C=O Vibration
The C=O stretching vibration is highly affected for intermolecular hydrogen atom. Hence, hydrogen bonding due to decrease the double bond character of the carbonyl group and shifting absorption band to lower frequency. The C=O stretching vibration is very strong and sharp band appearing in the region 1850-1550 cm-1 [31]. In presence of hydrogen atom near to the carbonyl group and it is simulated at 1723 cm-1 with 91% of PED. The theoretical wavenumber 1723 cm-1 also confirm that Gauss View animation option for given C17=O19.
C-C and C=C Vibrations
The ring C-C and C=C stretching vibrations usually occur in the regions 1650-1430 cm-1 and 1380-1280 cm-1, respectively. The actual positions of these modes are determined not so much by the nature of the substituent but rather by the form of the substitution around the ring [32]. According to C-C stretching vibrations are appears in region 1585, 1575, 1532 and 1476 cm-1 agree well with general range. The C=C stretching vibrations were established at 1339, 1284 and 1222 cm-1 which coincide with literature values. The in-plane and out-of-plane bends are reported at 652-509 cm-1 and 477-282 cm-1 respectively. The in-plane bending of βCCC is appear in the range 652 cm-1 with the PED percentage is 33%. The out-of plane δCCCC bend was observed at 419 and 204 cm-1.
C-Cl Vibration
The C-Cl stretching vibrations give generally strong bands in the region 800-600 cm−1. In the present peak at 652 cm-1 is agreed with literature value. The C–Cl deformation modes are appeared in the region 460–175 cm-1. In the present molecule peaks are appeared at 443, 410 and 228 cm-1 with PED percentage is 20, 42 and 39 % respectively.
C = N and C-N vibrations
The C-N stretching frequency is a very tough task since it falls in a composite region of the vibrational spectrum, i.e., mixing of several bands is possible in this region [33]. The C=N and C–N stretching modes appear around 1600–1500 cm−1 and 1300–1290 cm−1 [34] respectively. The C-N stretching vibration was observed at 1284 cm-1 which, is well matched with the literature value. The C=N stretching vibration is appeared in the region 1427 cm-1.
3.3. NMR spectral analysis
The isotropic chemical shifts are frequently used in identification of reactive organic as well as ionic species. It is recognized that accurate predictions of molecular geometries are essential for reliable calculations of magnetic properties [35]. The theoretical spectra were computed by gauge-independent atomic orbital (GIAO) [36] functional in combination with B3LYP method 6-31 G (d, p) basis set. The theoretical 1H and 13C NMR chemical shift values are presented in Table 3.
The computed 1H NMR chemical shift values are 10.4, 8.4, 8.2, 8.0, 7.8 and 7.7 ppm (18 –H, 14-H, 13-H,7-H,9-H and 10-H) respectively. Since, the proton numbered H18 has the maximum chemical shift value (10.4 ppm), as it is near the electronegative O19 atom. The protons 14-H, 13-H, 7-H, 9-H and 10-H with chemical shift values are 8.4, 8.2, 8.2 8.0, 7.8 and 7.7 ppm respectively. Whereas proton chemical shifts are due to shielding (or upfield). In the organic molecules, 13C NMR chemical shifts are usually lying in the region of 10-200 ppm [37]. In the present 13C NMR chemical shift values are 176.9, 138.7, 122.5, 116.0, 112.3 and 105.5 ppm (17-C, 12-C, 8-C, 1-C, 5-C and 11-C) respectively. The maximum chemical shift value of carbon atom 17-C having 176.9 ppm due to deshieled (downfield) and minimum chemical shift is 11-C with 105.5 ppm. The theoretical spectra of 13C NMR and 1H chemical shift are shown in Fig 3.
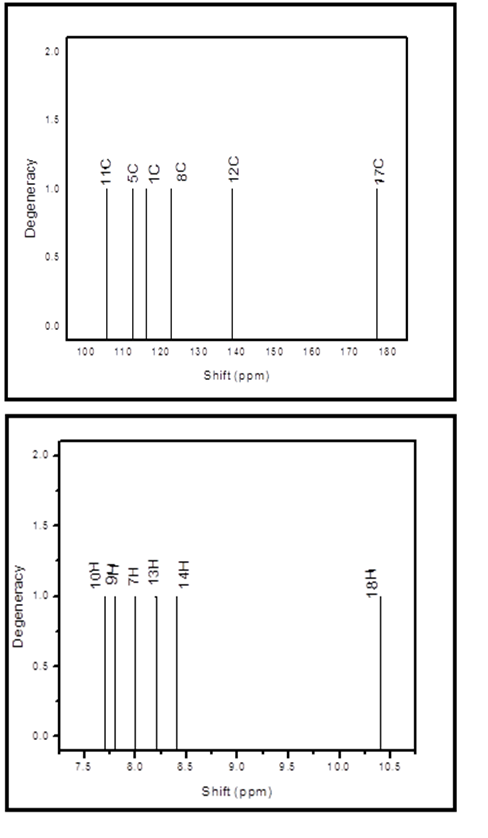
Fig 3. Theoretical spectra of 13C NMR and 1H NMR chemical shift of 8CQ2C.
Table 3. Theoretical chemical shift values of 13C NMR and 1H NMR
|
Atom |
Chemical Shift (ppm) |
Atom |
Chemical Shift (ppm) |
|
17-C |
176.9 |
18-H |
10.4 |
|
12-C |
138.7 |
14-H |
8.4 |
|
8-C |
122.5 |
13-H |
8.2 |
|
1-C |
116.0 |
7-H |
8.0 |
|
5-C |
112.3 |
9-H |
7.8 |
|
11-C |
105.5 |
10-H |
7.7 |
3.4. VCD spectral analysis
Vibrational circular dichroism (VCD) is similar to the electro-magneto-optic effect (Zeeman Effect) and it is directly related to IR of vibrational optical activity which detects differences in attenuation of left and right circularly polarized light passing through the compound. It is the extension of circular dichroism spectroscopy into the IR and near infrared region [38]. The VCD analysis were calculated by using DFT/B3LYP method with 6-31G (d,p) basis set. The theoretical VCD gives two dimensional structural information because VCD is sensitive to the mutual orientation of distinct ligand groups in a molecule. It is also used for the identification of absolute configurations of the organic compounds [39]. The vibrational difference with respect to the left and right circularly polarized light radiations and the result is combination of an emission and absorption spectra associated with biologically and optically significant of organic molecules.
In the present case, the VCD spectrum was originate from zero to 4000 cm-1 and the intensive peak started with 400 cm-1. The intense peaks are represented to the absorption and emission was predicted in both positive and negative phase due to the left and right polarization. The absorption intensity is usually unequal in both up and down of the VCD spectrum. The vibrational polarization bands belongs to middle IR region which corresponding to the C-C, C=C, C-N, C=N, C-Cl and C=O stretching respectively. These various vibrational modes are used to the optical and biological applications. The VCD spectrum is shown in Fig 4.
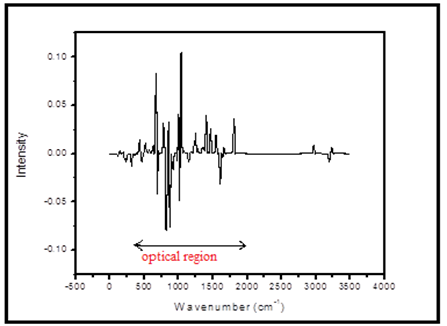
Fig 4. Theoretical VCD spectrum.
3.5. UV- Visible analysis
The theoretical UV- Visible spectrum were obtained by DFT/B3LYP method with 6-31G (d,p) basis set. The UV-Visible analysis was carried out different electronic transitions of the 8CQ2C. The calculated absorption maximum along with optical parameters such as excitation energy, excitation wavelength, and oscillator strength are given in the Table 4. The theoretical calculations predicted two peaks at 246.23 nm and 238.73 nm with corresponding excitation energies are 5.03 eV and 5.19 eV respectively. The maximum absorption peak at 246.23 nm are due to π →π* transition in the title compound. Theoretical UV- Visible spectrum is shown in Fig 5.
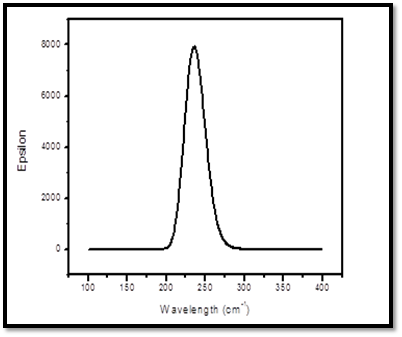
Fig 5. Theoretical UV- visible spectrum
Table 4. Theoretical electronic absorption spectrum values of 8CQ2C.
|
Excited states |
Wavelength (λmax) in nm |
Excitation energy (eV) |
Oscillator strength (f) |
Assignment |
|
S1 |
246.23 |
5.0353 |
0.1208 |
π →π* |
|
S2 |
238.73 |
5.1935 |
0.0586 |
π→π* |
3.6. Frontier molecular orbital energy analysis
HOMO means the highest occupied molecular orbital and LUMO means the lowest unoccupied molecular orbital. HOMO and LUMO are important parameters in defining the reactivity of chemical species [40-43]. The energy of HOMO indicates nucleophilicity and LUMO indicates electrophilicity [44]. HOMO-LUMO energy gap reflects the kinetic stability of the molecule [45]. The HOMO shows the various prominent donor orbitals and the LUMO shows that of prominent acceptor orbitals. HOMO−1 and LUMO+1, represents the respective donor and acceptor levels one energy state below and above these levels respectively [46]. The computed energy gaps and other quantum descriptors like electronegativity (χ), chemical hardness (η), softness(S), chemical potential (μ), and electrophilicity index (ω) are given in the title compound. HOMO-LUMO analysis were obtained by DFT/B3LYP method with 6-31G (d,p) basis set. The HOMO and LUMO orbitals of 8CQ2C are shown in Fig 6.
The electronic properties of the molecule are calculated from the total energies and the Koopman’s theorem [47]. The ionization potential (IP= - EHOMO), electron affinity (EA= -ELUMO), electrophilicity index (ω=μ2/2η), electronegativity (χ= (I+A)/2), chemical hardness (η= (I-A)/2) and softness (S=1/2η) are listed in Table 5. The calculated energy values of HOMO and LUMO are - 0.32668 and 0.03221 eV respectively. The energy gap between HOMO and LUMO is 0.29 eV. The values of IP, EA, χ are 0.3266eV, 0.0322 eV and 0.1472 eV. The chemical hardness is η = 0.1799 eV and softness is S =5.5574 eV were calculated in the title compound. The softness value of 8CQ2C indicates that it belongs to soft material category. The title compound of 8CQ2C is a negative ionization potential and it is stable molecule. The lowering of frontier molecular orbital energy gap confirms the charge transfer within the molecule which reflects its kinetic stability and substantiates its bioactivity.
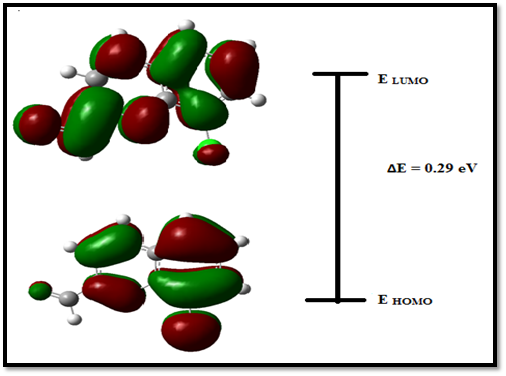
Fig 6. The HOMO and LUMO orbitals of 8CQ2C
Table 5. The Frontier Molecule Orbital values of 8CQ2C
|
FMOs |
8CQ2C |
|
EHOMO (eV) |
-0.3266 |
|
ELUMO (eV) |
-0.0322 |
|
EHOMO - ELUMO gap (eV) |
0.2944 |
|
Ionization Potential (I) |
0.3266 |
|
Electron affinity (A) |
0.0322 |
|
Electronegativity ( χ) |
0.1472 |
|
Chemical hardness (η) |
0.1799 |
|
Softness (S) |
5.5574 |
|
Electrophilicity index (ω) |
0.0601 |
3.7. Molecular Electrostatic Potential (MEP) analysis
MEP is very useful for docking analysis in the two species like protein and ligand interact mainly through their potentials. MEP surface helps to predict the reactivity of a wide variety of chemical systems in both electrophilic and nucleophilic reactions, the study of biological recognition processes and hydrogen bonding interactions [48]. The electrostatic potential increases in the order red < orange < yellow < green < blue [49]. The different values of the electrostatic potential at the surface are represented by different colors; red represents regions of most electro negative electrostatic potential, blue represents regions of most positive electrostatic potential and green represents regions of zero potential. MEP for the title compound is shown in Fig 7. The oxygen atom is present in 8CQ2C is surrounded by higher electron density given by deep red in the presence of double bond of carbon atom. The hydrogen atoms are attached in the carbon atoms and it is positive electronegative potential and the color representation is blue. The benzene ring is also zero potential in given color is green. The electrostatic potential is largely responsible for the binding of a substrate to its receptor binding sites since the receptor and the corresponding ligands recognize each other at their molecular surface [50, 51]. Hence, only the electrostatic potential of oxygen atom is interact with two amino acids like SER A and TYR A in the active side of the ligand.
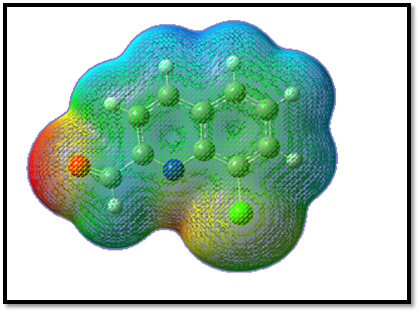
Fig 7. Molecular Electrostatic Potential of 8CQ2C
3.8. Natural bonding orbital (NBO) analysis
NBO analysis is performed to determine the electronic transitions from filled orbital of one system to unfilled orbital of another system. The second order fock matrix method is carried out to the interactions of donor-acceptor in the NBO analysis [52]. NBO analysis was calculated by using DFT/B3LYP method with 6-31G (d,p) basis set. It is mainly used to measure the delocalization of electron density and such a kind of hyper conjugation of electronic orbitals. The higher the E(2) values, the molecular interaction of donor – acceptor are more intense and greater is the stability of entire molecule. The analysis of various donors and acceptors are indicates only two types of donors π & σ and two types of acceptors π *& σ* respectively.
The Delocalization of electron density amid occupied Lewis-type (bond or lone pair) NBO orbitals and properly unoccupied (antibond or Rydgberg) non-Lewis NBO orbitals resemble to a stabilizing donor-acceptor interaction. The second order perturbation of stabilization energies E(2) between bonding and antibonding has been given in the Table 6. The maximum stabilization energy is 23.67 kJmol-1 and it is obtained for the interactions between π (C8-C11) → π * (C12-N16). In the other interactions are LP(2)O19 → π * (C17-H18), π(C12-N16) → π * (C3-C4), π (C3-C4) → π * (C12-N16), π (C3-C4) →π * (C1-C2) and π (C8-C11) → π * (C3-C4) which leads to strong delocalization of 20.78, 20.04, 16.44, 16.05 and 16.02 respectively. The very important interactions of Lewis bond and non-Lewis bond orbitals with the Cl15, N16 and O19 lone pairs. The maximum intermolecular charge transfer are occurs in the π bonding and π * antibonding orbitals thus the entire title compound have a π conjugated system and it is more stability. The more reactive side is carbonyl group (C17=O19) and it also interacts with good way of protein-ligand interactions.
Table 6. NBO transition data of 8CQ2C calculated using second order perturbation method of Fock matrix.
|
Donor (i) |
ED(e) |
Acceptor (j) |
ED(e)
|
E(2)a Kal/Mol |
E (i) – E(j)b a.u |
F(i,j)c |
|
π (C3-C4) |
1.96632 |
π * (C1-C2) |
0.29982 |
16.05 |
0.27 |
0.062 |
|
π (C3-C4) |
1.51545 |
π * (C12-N16) |
0.34209 |
16.44 |
0.26 |
0.061 |
|
π (C8-C11) |
1.98090 |
π * (C3-C4) |
0.46074 |
16.02 |
0.28 |
0.063 |
|
π (C8-C11) |
1.70976 |
π * (C12-N16) |
0.01796 |
23.67 |
0.28 |
0.073 |
|
π (C12-N16) |
1.98528 |
π * (C3-C4) |
0.04219 |
20.04 |
0.32 |
0.076 |
|
LP(3)Cl15 |
1.91820 |
π * (C1-C2) |
0.02281 |
13.88 |
0.33 |
0.064 |
|
LP(1)N16 |
1.91688 |
π * (C11-C12) |
0.03637 |
11.13 |
0.87 |
0.089 |
|
LP(2)O19 |
1.88672 |
π * (C17-H18) |
0.05905 |
20.78 |
0.69 |
0.108 |
ED, Electron density,
aE(2) means energy of hyper conjugative interaction (stabilization energy),
bEnergy difference between donor and acceptor i and j NBO orbitals,
cF (i, j) Fork matrix element between i and j NBO orbitals.
3.9. Mulliken atomic charges
The Mulliken population analysis gives the individual charge on each atoms in the molecule and it is the several characteristics of the molecular structure. The mulliken atomic charges were obtained by DFT/B3LYP method with 6-31 G (d, p) basis set. The mulliken atomic charges are shown in Fig 8. Among the carbon atoms, all the carbon atoms except C3, C4, C6, C12 and C17 are all of negative charges. The presence of nitrogen and oxygen atoms are more electronegative and it makes the two double bond of carbon atoms C12 and C17 more negative. Hence N16 and O19 are more negative which can attract the more atoms from the neighbor. The Cl15 atom is one of positive charge attached to the electronegative atom C2 because this atom makes the double bond C2 and C1 atoms are the electro negativity. All the hydrogen atoms are positively charged because they donate electrons to the nearby carbon atoms [53] and all the carbon and nitrogen atoms are negatively charged. The atomic charges are given in the Table 7. Hence, the sums of Mulliken charges of all atoms are calculated to be zero and confirmed the charge neutrality.
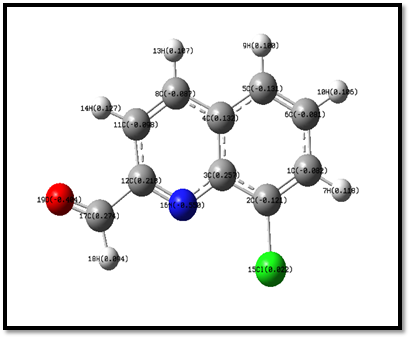
Fig 8. Mulliken atomic charges.
Table 7. Mulliken atomic charges of 8CQ2C
|
Atomic Number |
Natural atomic charges |
|
C1 |
-0.082 |
|
C2 |
-0.121 |
|
C3 |
0.257 |
|
C4 |
0.132 |
|
C5 |
-0.131 |
|
C6 |
0.081 |
|
H7 |
0.118 |
|
C8 |
-0.087 |
|
H9 |
0.100 |
|
H10 |
0.106 |
|
C11 |
-0.098 |
|
C12 |
0.218 |
|
H13 |
0.107 |
|
H14 |
0.127 |
|
Cl15 |
0.022 |
|
N16 |
-0.550 |
|
C17 |
0.274 |
|
H18 |
0.094 |
|
O19 |
-0.404 |
3.10. Non-Linear Optical (NLO) Property
The NLO activity provides the key functions for optical modulation and switching, frequency shifting and optical logic for the developing technologies in areas such as communication, signal processing and optical inter connections [54,55]. Organic molecules able to manipulate photonic signals efficiently are of importance in technologies such as optical communication, optical computing, and dynamic image processing [56,57]. The polarizability, hyperpolarizability and dipole moment also calculated by using DFT/B3LYP method with 6-31 G (d,p) basis set. To find out the dipole moment (μ), polarizability (α), and hyper polarizability (β) are defined as [58] using x, y and z components,
Dipole moment is
μ=(μ2x+μ2y+μ2z)½
Polarizability is
α0=(αxx+αyy+αzz)/3
Hyperpolarizability is
β=(β2x+β2y+β2z)½
Where
βx = (βxxx +βxyy +βxzz)
βy = (βyyy +βyzz+ βyxx)
βz = (βzzz +βzxx +βzyy )
β = [(βxxx + βxyy + βxzz)2 + (βyyy +βyzz +βyxx)2 + (βzzz +βzxx +βzyy)2]1/2
The polarizability and hyperpolarizability values are given by Gaussian 09 and it is reported in atomic units (a.u), the calculated values have been converted into electrostatic units (e.s.u) (α ; 1 a.u. = 0.1482x 10-24 e.s.u; β; 1 a.u.= 8.3693 x 10-33 e.s.u) [59]. The values of dipole moment is μ = 4.885 D and hyperpolarizability is β = 6.7349 x 10-30 esu. The title compound is 18 times greater than that of the standard NLO material Urea (0.3728 x 10-30 esu). Hence, 8CQ2C also has NLO property. The calculated β components and βtot value of 8CQ2C are given in Table 8.
Table 8. Calculated β components and βtot value of 8CQ2C
|
β Components |
8CQ2C |
|
βxxx |
521.3285 |
|
β xxy |
224.6322 |
|
βxyy |
13.76503 |
|
β yyy |
220.9898 |
|
βxxz |
14.46556 |
|
βxyz |
8.52040 |
|
βyyz |
-5.54994 |
|
βxxz |
-0.02040 |
|
βyzz |
-0.39329 |
|
βzzz |
0.03289 |
|
βTotal (esu) |
6.73495 X 10-30 |
3.11. Molecular docking analysis
The molecular docking method is used to find out the ligand binding site to a receptor and predict the binding orientations. Now a days, computational investigations act as emerging tools for studying a number of molecular parameters [60, 61]. In the present work, the molecular docking analysis was carried out for the 8CQ2C ligand with more common targeted proteins associated with the Parkinson’s and schizophrenia diseases. Parkinson’s disease is a neurodegenerative disorder and Schizophrenia is a brain disorder but it also life –long disease that cannot be cured but can be controlled with proper treatment. Recently, many researchers have been focusing the molecular docking studies on the mentioned targeted proteins [62-65].
To find the two proteins like 2V60 and 4MRW and these structures of the targeted proteins were obtained from the RCSB PDB format [66]. Autodock Tools version 1.5.6 is recently have been used as a convenient tool to get insights of the molecular mechanism of ligand-protein interactions and to bind the receptor of 3D structure. Discovery studio visualizer were utilize for the evaluation of hydrogen bonds in the ligand-protein interaction. The ligand PDB file was created by using the optimized molecular structure of the 8CQ2C molecule. The AutoDock Tools graphical user interface [67] was used to prepare the target proteins for docking. The ligand and water molecules present in the targeted proteins were removed. The polar hydrogen bond and Kollman charges were added in the targeted proteins.
The intermolecular interactions were studied for the 8CQ2C with different proteins like 2V60 and 4MRW. As a result of docking is find out the ten conformers were obtained and their binding energies with inhibitory constants were listed in given Table 9. The binding energies with inhibitory constants of 2V60 and 4MRW are found to be -6.76 and -5.7 k cal mol -1 with 11.1 and 6.91 um respectively. 3D model interaction of title compound with proteins shown in Fig 9. 2D model of interaction between amino acid residues is shown in Fig 11 and it is predicted the green dotted lines are represent the hydrogen bonds, the dark yellow dotted lines re represent the electrostatic bonds and pink dotted lines are represent the hydrophobic bonds. The amino acids are mainly involved in the ligand and protein interactions. The docking of 8CQ2C interact with different amino acids like Serine A: 59 with 2.10 Å (Symbol SER A: 59) and Tyrosine A:60 with 2.25 Å (Symbol TYR A:60) in 2V60 protein. For another amino acid like Tyrosine A: 524 with 2.61 Å (Symbol TYR A: 524) were interacted with 8CQ2C and 4MRW protein. These interactions are depends upon the nature of the functional groups present in the ligand. The docked confirmation of the active site of 8CQ2C is shown in Fig 10. These results indicate that the 8CQ2C ligand possesses the lowest binding energy and inhibition constant for the targeted proteins. Hence, these docking results will be useful for developing the effective treatment of Parkinson’s and Schizophrenia diseases.
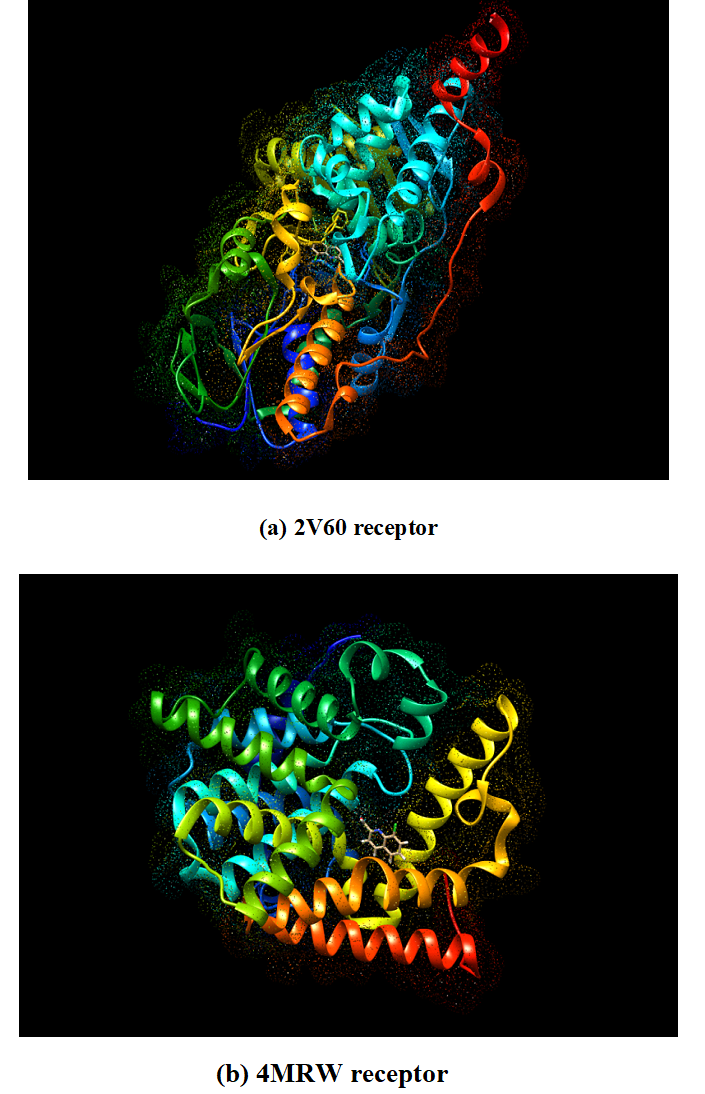
Fig 9. 3D Model of ligand 8CQ2C with (a) 2V60 and (b) 4MRW receptors
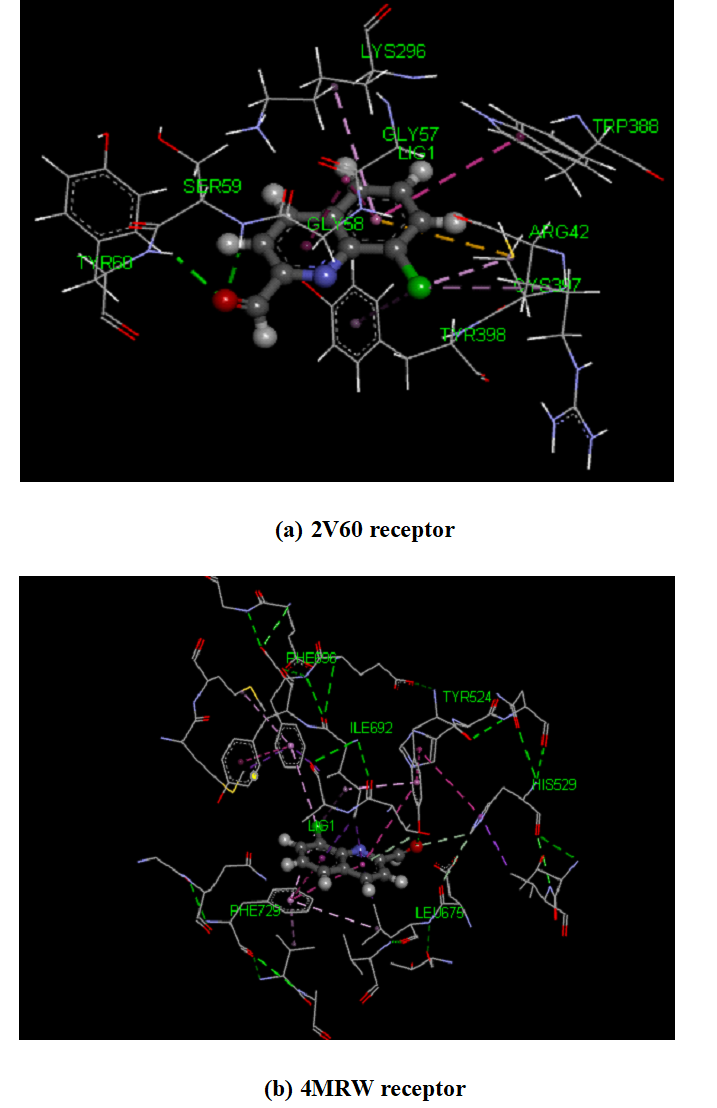
Fig 10. The schematic of the docked confirmation of the active site of 8CQ2C with (a) 2V60 and (b) 4MRW receptors.
Table 9. The possible conformers of 8CQ2C docked with 2V60 and 4MRW receptors.
|
Conformer |
Binding Energy(k cal mol-1) |
Inhibitory constant(μM) |
||
|
2V60 |
4MRW |
2V60 |
4MRW |
|
|
1. 2. 3. 4. 5. 6. 7. 8. 9. 10. |
-6.76 -6.76 -6.76 -6.75 -6.76 -6.75 -6.76 -6.76 -6.75 -6.76
|
-5.7 -5.7 -5.59 -5.59 -5.69 -5.7 -5.69 -5.68 -5.6 -5.7 |
11.1 11.11 11.16 11.24 11.07 11.33 11.13 11.04 11.2 11.12 |
66.58 66.42 79.3 80.36 67.27 66.91 67.34 68.41 79.09 66.69
|
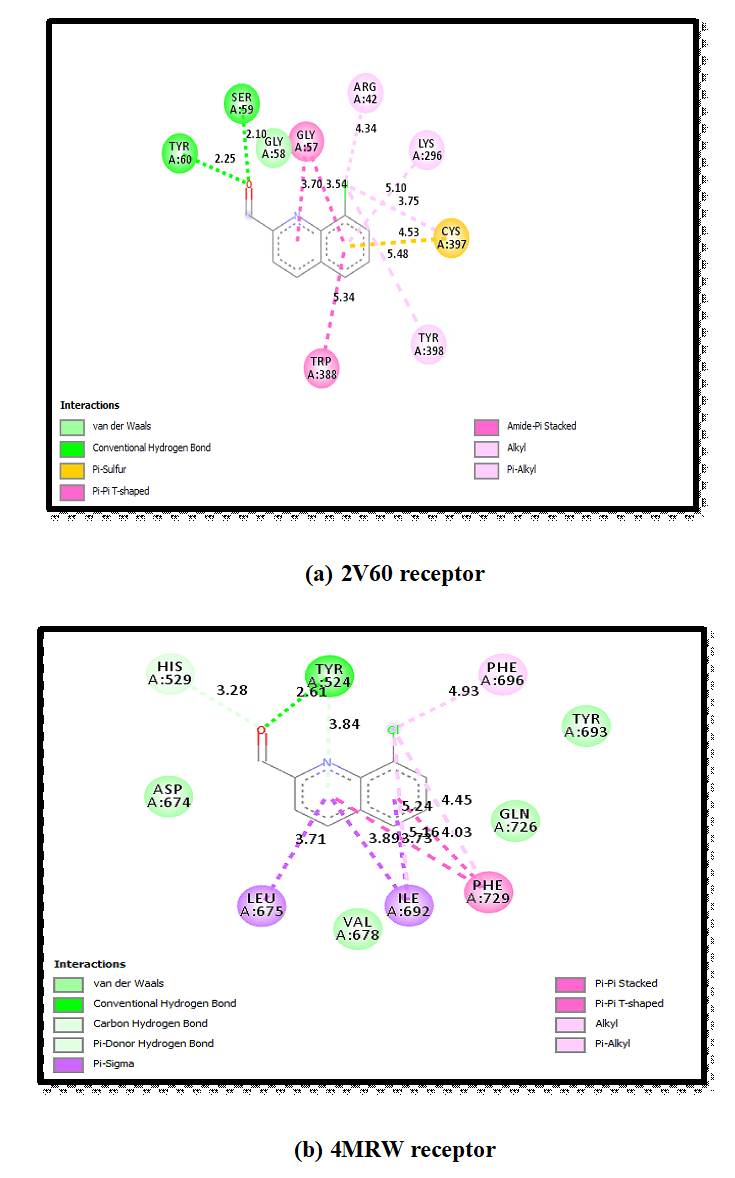
Fig 11. 2D model of interaction between amino acid residues with 8CQ2C and 2V60 & 4MRW.
In the present work, 8CQ2C were optimized by using DFT/B3LYP method with 6-31 G (d,p) basis set. The optimized structure provides various bond lengths and bond angles coinciding with the literature values and also calculated the global minimum energy E= - 974.8020 Hartrees in title compound. The various vibrational frequencies are calculated on the basis of PED by using VEDA program. The theoretical NMR provides the 1H NMR and 1C NMR chemical shift values are reported. The theoretical VCD spectrum was simulated and also predicted to both obtain optical and biological activity of the title compound. The UV-Vis analysis was simulated theoretically and the maximum absorption peak is 246.23 nm is associated with π →π* transition. The band gap energy is 0.29 eV and the HOMO-LUMO provides the energy difference between the charge transfer interactions in the molecule. The possible electrophilic and nucleophilic reactive sites were predicted by MEP. In the negative potential sites are on oxygen, chlorine and nitrogen atoms as well as the positive potential sites are hydrogen atoms in the title compound. Stabilization of title compound is carried out by hyper conjugation interactions and charge delocalization has been calculated by NBO analysis. Mulliken charge distribution has been calculated. The dipole moment, polarizability and hyperpolarizability reveal that title compound has considerable NLO activity and hence can be used for material science applications. The docking result clearly provides that the two different proteins interact with ligand. The most active side is C17=O19 of 8CQ2C ligand interactions takes for different amino acids like SER A: 59, TYR A: 60 and TYR A: 524 with given resolutions 2.10 Å, 2.25 Å and 2.61 Å respectively. Hence, 8CQ2C may be used in the treatment of Parkinson’s and Schizophrenia diseases but in-vito & in-vivo studies have to been carried out before implementation.
The authors gratefully acknowledge the support of this work by Department of Physics, Sri Sarada College for Women (Autonomous), Salem-16.