Kafoumba BAMBA
Email: bambakaf.sfa@univ-na.ci
© 2019 Sift Desk Journals. All Rights Reserved
VOLUME: 4 ISSUE: 1
Page No: 313-320
Kafoumba BAMBA
Email: bambakaf.sfa@univ-na.ci
Fatogoma DIARRASSOUBA1, Kafoumba BAMBA*1, Mawa KONÉ*2, Ahissan Donatien EHOUMAN1, Sopi Thomas AFFI1
1Laboratoire de Thermodynamique et de Physico-chimie du Milieu, UFR-SFA, Université Nangui Abrogoua, 02 BP 801 Abidjan 02, République de Côte-d’Ivoire
2Laboratoire de Chimie Organique et des Substances Naturelles, UFR-SSMT, Université Félix Houphouët Boigny, 22 BP 582 Abidjan 22, République de Côte-d’Ivoire
Beatriz Sevilla-Morán(bsmoran@inia.es)
BAMBA Kafoumba, Design of new electron acceptors based on Tetracyanoquinodimethane (TCNQ) by molecular modeling techniques and Structure-Property Relationship (SPR) study(2020)Journal of Computational Chemistry & Molecular Modeling 4(1)
In our previous work, (Fatogoma D., Mawa K., Kafoumba B., Yafigui T., Mamadou G.-R. K., Edja F. A., Computational Chemistry, 7(4), 121-142, 2019) we established a QSPR (Quantitative Structure-Property Relationship) model of the first reduction potential which depends on electronic affinity (EA) only. It has been reported that this model is suitable for the prediction of the first reduction potential of new Tetracyanoquinodimethane (TCNQ) acceptors belonging to its applicability domain with a 95% confidence level. The present work, which is a continuation of the previous, has the main objective to design new molecules of Tetracyanoquinodimethane (TCNQ) belonging to the applicability domain of this model and to predict their first reduction potentials. In this context, we have designed a series of sixty (60) new molecules of Tetracyanoquinodimethane (TCNQ). All these molecules have been optimized at the B3LYP/6-31G (d,p) theory level. The values of the first reduction potential of these molecules calculated by using the model, revealed they have moderate oxidizing powers since the values of their potentials are between -0.02 V and +0.35 V (Robert C. Wheland, 98(13), 3926-3930, J. Amer. Chem. Soc., 1976). This means that they are electronically able to form organic conductive salts. In addition, it has been found that positional isomers have practically the same first reduction potential. Globally, the electro-donor functions attenuate the oxidizing power of the molecules when the electro-attractor functions increase the oxidizing power. In view of the thermodynamic quantities, the formation reaction of these new electron acceptors is spontaneous with release of heat and decrease of the disorder. All electro-donor groups are stabilizing. On the other hand, except for the chloro function, all the remaining electro-acceptor functions are stabilizing notably the cyano group. The new acceptors designed in this work are potential candidates for organic electronics domain.
Keywords: Tetracyanoquinodimethane (TCNQ), first reduction potential, oxidizing power
The 7,7,8,8-Tetracyano-p-quinodimethane [1] or more simply Tetracyanoquinodimethane (TCNQ) [2] has been the subject of frequent experimental and theoretical research [3,4] since the discovery of electrical conductivity salts that form the radical anion TCNQ.- with organic cations. The development of plane geometry electron acceptors and delocalized π-electron systems is a prerequisite in the development of charge transfer complexes [5].
Tetracyanoquinodimethane (TCNQ) is a strong electron acceptor with a high electronic affinity ranging from 2.8 eV [6,7] to 3.383 eV [8] due to the presence of cyano groups and the possibility of electron conjugation of the π system [9]. It is easily reduced to a radical anion (TCNQ.-) then dianion (TCNQ2-). The substitution of hydrogen atoms by electro-donor groups (alkyl and alkoxy) or electro-attractor (halogen and cyano) causes a variation of the energy barrier. By adding these groupings, it is possible to reduce the energy barrier of the forbidden band, bringing the HOMO (Highest Occupied Molecular Orbital) and the LUMO (Lowest Unoccupied Molecular Orbital) closer to each other.
For example, the electro-attractor groups on a conjugated molecule have the effect of attracting the electron density towards them [10,11]. They are able to stabilize a large electron density. As a result, the rest of the molecule becomes depleted of electrons, which lowers the energy of HOMO (Highest Occupied Molecular Orbital).
The main objective of this work is to exploit the predictive QSPR (Quantitative Structure-Property Relationship) model of the first reduction potential of Tetracyanoquinodimethane (TCNQ) developed in our previous work (F. Diarrassouba et al. [12]). In fact, it is clearly a question of designing a series of new analogous Tetracyanoquinodimethane (TCNQ) molecules belonging to the applicability domain of this model that will electronically be capable of complexing with electron donors to form organic electrical conductive salts.
2.1. Structure of the reference molecules
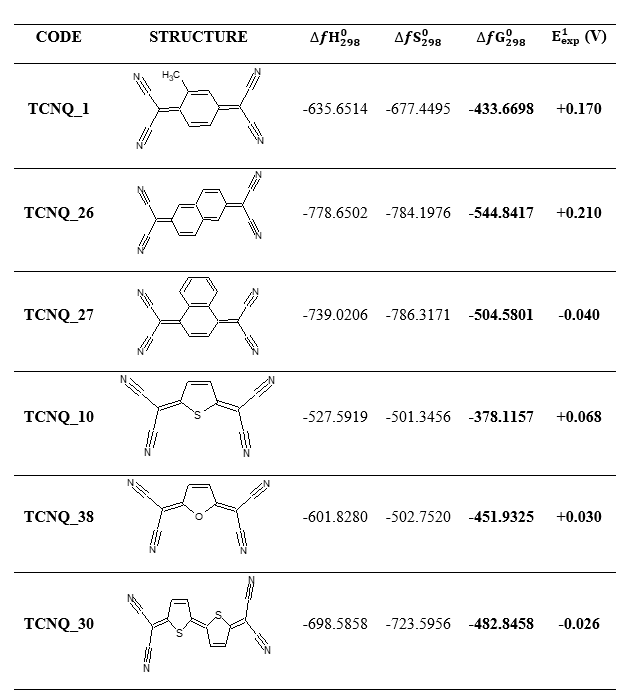
The new molecules were designed by replacing the hydrogen atoms of the reference compounds of our experimental database [12] with electro-donor and electro-acceptor functions. The structures of these molecules as well as their first reduction potentials are summarized in Table 1. Regarding the thermodynamic quantities of the formation, they have been calculated in the present work in order to understand the evolution of the thermodynamic stability and the property (first reduction potential) in function of the introduced groups.
Table 1. Structures of reference TCNQ compounds
2.2. Computational Theory Level and Software’s
The gaussView 5.0 [13] software was used to represent the 3D structure and visualize the studied molecules. Then, the Gaussian 09 software [14] was used for optimization and frequency calculation at the temperature of 298.15 Kevin, 1 atmosphere as pressure and in vacuum. The theory level used is B3LYP/6-31G(d,p). As for 2D structures, they have been represented with chemsketch [15].
2.3. Fragment method [16]
For the prediction of the properties of new compounds, the fragment method is qualitative. When a compound contains only fragments of the compounds of the database, the prediction of its property is reliable. On the other hand, if the compound contains unknown fragments of the database, the prediction is then doubtful.
2.4. Min-Max method [16]
The Min-Max method is based on the minimum (dmin) and maximum (dmax) values whose interval is denoted D=[dmin-dmax] for each descriptor. Only the values of the descriptors of the compounds of the training set are considered. The prediction of the property of a new compound is reliable only when the value(s) of its descriptor(s) belongs to the corresponding interval D. Otherwise, the prediction will be doubtful.
After ranking in ascending the order of the values of the only descriptor of the compounds of the training set [12], it follows that eV
2.5. QSPR model used to predict the first reduction potential
The QSPR model used for the design of the new electron acceptors was developed in our previous work [12]. The equation of this model is the following with its statistical parameters and validations below:
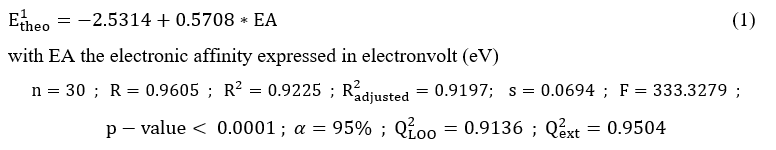
2.6. Standard thermodynamic formation quantity
The results of the quantum calculation and the use of experimental data in the form of reference thermodynamic tables [17] make it possible to determine the standard values of the enthalpy ∆f H0, the entropy ∆f S0 and the free enthalpy ∆f G0 of formation of the molecular system at 298.15 K under a pressure of 1 atm.
The calculation of molecular system enthalpy of formation is carried out according to the relation of J. W. Ochterski [18]:
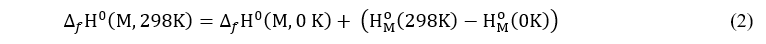
where the last term of this relation is called enthalpy correction or increment. For simple elements, it corresponds to the heat involved during the heating of the atoms from 0 K to 298 K. These values are obtained experimentally relative to the standard state of the elements. As for the term ∆f H0(M,0 K) , it is determined from the following relation:
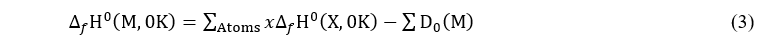
x defines the number of atoms of species X in molecule M
∆f H0(X,0 K)is an experimental value corresponding to the heat of formation of the atom X at 0 K.
The last term ∑D0(M), ,corresponds to the atomization energy of the molecule M. This energy comes from the decomposition of one mole of the molecule into its constituent atoms. The calculation of ∑D0(M) is performed from the electronic energies (ε) of the constituent atoms and the molecule and the internal energy at 0 K called ZPE (Zero-Point Energy) of the latter according to the relation below:
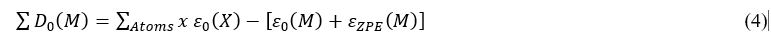
The determination of the formation entropy at 298 K, ∆f S0(298K) is made using the atomic entropy values S0(X,298K) from JANAF Thermochemical Tables [17] and the molecular entropy obtained by quantum computation. The proposed relation by J. W. Ochterski [18] for determining the entropy of formation of a molecular system is:
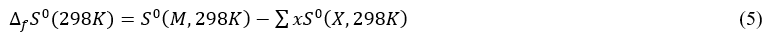
Knowing the enthalpy of formation and entropy of formation, the free standard enthalpy of formation is easily calculated by the relation:
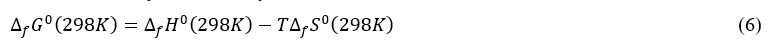
with T=298,15 K
3.1. Design of new electron acceptors based on tetracyanoquinodimethane (TCNQ) and Structure-Property Relationship (SPR) study
The QSPR model was used to predict the first reduction potential of a series of sixty (60) novel designed tetracyanoquinodimethane molecules codified HA_i with all belonging to its applicability domain. The structures as well as the different quantities are summarized in Table 2.
Table 2. Structures of the molecules with electronic affinities, enthalpy and free enthalpy of formation (in kcal/mol), entropy of formation (in cal/mol.K) and predicted values of the first reduction potentials by the model
https://www.siftdesk.org/articles/images/10599/t2.pdf
It is noted in Table 2 that the electronic affinity (EA) values of the new compounds all belong to the interval D, which translates into reliable prediction. Also, the comprehensive designed Tetracyanoquinodimethane molecules have moderate oxidizing powers as all theoretical first reduction potentials are between -0.02 V and + 0.35 V (-0.02V ≤ E1theo ≤ +0.35V) [19].
By comparing the redox potentials of the parent molecules (Table 1) with those of the new molecules, it appears that the "grafting" of the different functions on the parent molecules has an influence on the redox potential. In the series of new molecules, positional isomers have virtually the same redox potentials. The position of the functions on the parent molecules therefore has no influence on the studied property. However, increasing these functions on any molecule in the series influences the redox potential. Overall, it is found that the oxidizing power of molecules increases with the increase in the number of electro-attractors functions while it decreases as the number of electro-donor functions increases. In the case of the electro-attractor functions, the oxidizing power is more accentuated with the increase of the number of cyano function unlike the fluoro and chloro function. This observation is explained by the fact that the cyano group has a very attractive mesomeric effect, more important than an attracting inductive effect. When the electro-donor functions, the opposite case is observed. The electro-donor groups increase the stability of the molecules but attenuate the oxidizing power. Under these conditions, the very strong oxidizing power of a molecule can be attenuated by "grafting" the electro-donor functions. Also, the values of the standard thermodynamic quantities of formation of the new molecules are all negative. Negative values of enthalpy and free enthalpy indicate respectively an exothermic reaction and a spontaneous reaction. A negative value of the entropy indicates a decrease in the disorder. As a result, the formation of all new electron acceptors is spontaneous with heat release and disorder reduction under the temperature conditions of 298.15 K under 1 atm pressure in the vacuum. The negative value of the free enthalpy of formation thus reflects the existence and stability of new TCNQ molecules. Furthermore, the standard free enthalpy of formation decreases with the gradual introduction of the fluorine atom while it increases with the increase in the number of chlorine atoms.
Thus, the progressive addition of fluorine increases the thermodynamic stability of the molecules when it decreases with the gradual introduction of chlorine atom. The chlorine atom increases the oxidizing power of TCNQ faster than fluorine atom, but the latter stabilizes better thermodynamically. As these molecules are all new, it would be wise in the following to verify if this same conclusion is made for halogenated derivatives, cyano and methoxy of the experimental database.
3.2. Structure-Property Relationship (SPR) study of the halogenated derivatives, methoxy, cyano of the experimental database
To verify the consistency of our theoretical results, we studied the evolution of the redox property and the stability of halogenated derivatives, cyano and methoxy of our experimental database [12]. The selected structures are represented in Table 3.
Table 3. Structures, electronic affinities, theoretical enthalpy and free enthalpy (in kcal/mol) entropy of formation (in cal/mol.K) and experimental values of the first reduction potentials of the halogenated derivatives, cyano and methoxy of the experimental database.
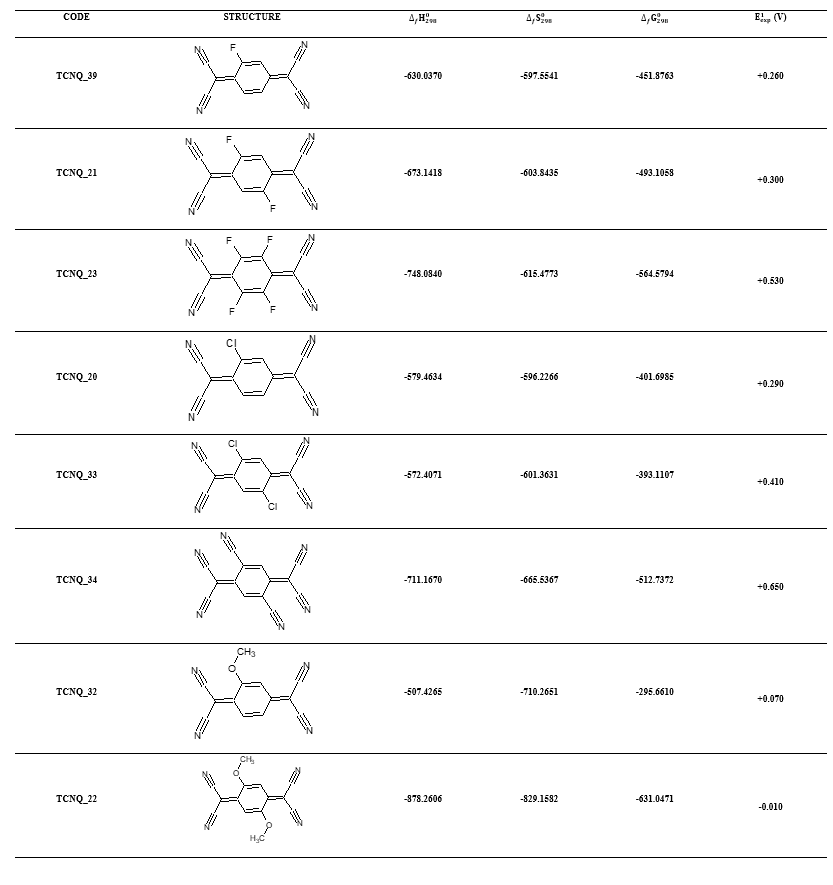
Globally, all electro-attractor groups increase the first reduction potential while the electro-donor groups attenuate the first reduction potential. In the case of the electro-attractor groups, starting from the molecules having the same numbers of identical substituents, it is noted that with the cyano group, the redox potential increases more rapidly. Regarding the chloro function, it increases the redox potential faster than the fluoro function. This is the case for TCNQ_21, TCNQ_22 TCNQ_33. In terms of thermodynamic stability, apart from chlorinated compounds, all the other compounds are stabilizing irrespective of the nature of the "grafted" function. However, the stability is more accentuated with the cyano group. Fluorine is more stabilizing than chlorine but it increases the redox potential more than fluorine. Hence, we exactly obtain the same trend as the theoretical results obtained during our work. This proves the consistency and reliability of our theoretical results as well as the performance of our developed QSPR model in our previous work. The electron acceptors designed in this work will electronically be capable of complexing with electron donors including tetrathiafulvalene (TTF) to form organic electrically conductive salts. They are therefore very promising for the field of organic electronics.
The sixty (60) electron acceptors molecules designed for this work have all moderate oxidative powers as their first reduction potentials are between -0.02 V and + 0.35V. The formation of each of these different molecules is spontaneous under the temperature conditions 298.15 K and under a pressure of 1 atm in the vacuum as the free enthalpies of formation are negative. Also, the formation reactions of these molecules are done with release of heat and decrease of disorder. The electro-donor functions are stabilizing by inductive effect but attenuate the oxidizing power of the molecules. On the other hand, the electro-attractor functions increase the oxidative power of the TCNQ but the oxidizing power is more pronounced for the cyano group. Regarding chlorine, it increases the oxidizing power more than the fluorine. In terms of thermodynamic stability, the cyano group and the fluorine stabilize the molecules but the cyano group is more stabilizing. On the other hand, the "grafting" of chlorine on the molecules increases the free enthalpy of formation. This leads to a destabilization of the molecules. Therefore, we can assume that the new designed acceptors are potential candidates for the field of organic molecular electronics. Thus, they are very promising for developing future conductive organic materials.
L.R. Melby, R.J. Harder, W.R. Hertler, W. Mahler, R.E. Benson et W.E. Mochel, Substituted Quinodimethans. 11. Anion-radical Derivatives and Complexes of 7,7,8,8-Tetracyanoquinodimethan, J. Am. Chern. Soc., , 84(17), 3374-3387, 1962
View ArticleHsu, C.-L., Lin, C.-T., Huang, J.-H., Chu, C.-W., Wei, K.-H., & Li, L.-J.. Layer-by-Layer Graphene/TCNQ Stacked Films as Conducting Anodes for Organic Solar Cells,. ACS Nano, 6(6), 5031-5039, 2012 PMid:22632158
View Article PubMed/NCBIV. G. Zakrzewski, O. Dolgounitcheva, and J. V. Ortiz, Electron binding energies of TCNQ and TCNE, J. Chem. Phys., 105(14), 5872-5877, 1996
View ArticleJ. B. Torrance, The Difference between Metallic and Insulating Salts of Tetracyanoquinodimethane (TCNQ): How to Design an Organic Metal, Acc. Chem. Res. 12(3), 79-86, 1979
View ArticleJames J. Delaney B.Sc., Synthesis of New Heterocyclic TCNQ Analogues, Doctorate of Philosophy, Dublin City University (School of Chemical Sciences), 202 p, 1997.
Klots, C. E., Compton, R. N. & Raaen, V. F. Electronic and ionic properties of molecular TTF and TCNQ, J. Chem. Phys. 60(3), 1177-1178, 1974
View ArticleMilián, B., Pou-Amérigo, R., Viruela, R. & Ortí, E. On the electron affinity of TCNQ, Chem. Phys. Lett. 391(1-3), 148-151, 2004
View ArticleZhu, G.-Z. & Wang, L.-S. Communication: Vibrationally resolved photoelectron spectroscopy of the tetracyanoquinodimethane (TCNQ) anion and accurate determination of the electron affinity of TCNQ, J. Chem. Phys.,143(22), 221102-4, 2015 PMid:26671350
View Article PubMed/NCBIYui, K., Aso, Y., Otsubo, T., & Ogura, F., Extensively Conjugated Homologues of Selenophene-TCNQ as New Electron Acceptors, Chem. Lett., 17(7), 1179-1182, 1988
View ArticleJiao C., Huang K.-W., Luo J., Zhang K. , Chi C., Wu J., Bis-N-annulated Quaterrylenebis(dicarboximide) as a New Soluble and Stable Near-Infrared, Dye, Organic Letters, 11(20), 4508-4511, 2009 PMid:19772312
View Article PubMed/NCBINetzeva, T.I., Worth A. P., Aldenberg T., Benigni R., Cronin M. T., Gramatica P., Jaworska J.S., Kahn S., Kloman G., Marchant C. A. and Myatt, G., Current status of methods for defining the applicability domain of (quantitative) structure-activity relationships, ATLA, 33(2),155-173, 2005 PMid:16180989
View Article PubMed/NCBIFatogoma D., Mawa K., Kafoumba B., Yafigui T., Mamadou G.-R. KONÉ, Edja F. A., Development of predictive QSPR model of the first reduction potential from a series of Tetracyanoquinodimethane (TCNQ) molecules by the DFT (Density Functional Theory) method, Computational Chemistry, 7(4), 121-142, 2019
View ArticleR Dennington, T Keith and J Millam., GaussView Version 5, Semichem Inc., Shawnee Mission, KS, 2009.
Gaussian 09, Revision A.02, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian, Inc., Wallingfor d CT, 2009.
ACDLABS 10, Advanced Chemistry Development Inc., Toronto, ON, Canada, 2015.
Netzeva, T.I., Worth, A.P., Aldenberg, T., Benigni, R., Cronin, M.T., Gramatica, P., Jaworska, J.S., Kahn, S., Kloman, G., Marchant, C.A. and Myatt, G., Current status of methods for defining the applicability domain of (quantitative) structure-activity relationships, ATLA, 33, 155-173, 2005 PMid:16180989
View Article PubMed/NCBIM. W. Chase, Jr. , C. A. Davies, J. R. Downey, Jr., D. J. Frurip, R. A. McDonald, and A. N. Syverud, J. Phys. Ref. Data 14 Suppl. No. 1; (JANAF Thermochemical Tables), 1985
Joseph W. Ochterski, Thermochemistry in Gaussian, help@gaussian.com©2000, Gaussian, Inc., June 2, 2000
R.C. Wheland, Correlation of Electrical Conductivity in Charge-Transfer Complexes with Redox Potentials, Steric Factors, and Heavy Atom Effects, J. Amer. Chem. Soc.,98(13), 3926-3930, 1976.
View Article