Said Ghalem
Email:s_ghalem2002@yahoo.fr
Molecular docking of 11β-HSD1 with (1,2,4-triazole derivatives) inhibitors in a solvated medium
Corresponding Author
Affiliation
MostefaouiLarbi, MeriemMerad, benmiloudkamal and Said Ghalem*
Laboratory of Naturals Products and Bio actives-LASNABIO
Faculty of Science, Department of Chemistry, University of Tlemcen, Tlemcen, Algeria
Citation
M Larbi, M Merad, B kamal and S Ghalem, Molecular docking of 11?-HSD1 with (1,2,4-triazole derivatives) inhibitors in a solvated medium(-0001)SDRP Journal of Computational Chemistry & Molecular Modelling 2(2)
Abstract
Background: The scope of molecular modeling techniques is enormous, reflecting the spectrum of knowledge and scientific domains that may be involved. It can be valuable in the identification of therapeutic targets, typically protein receptors whose specific role in a pathology is characterized. It may then prove indispensable in the development of chemical inhibitors for this target.1, 2, 4 triazol derivatives are recommended in treatment of hypertension.
Methods:The study of the interaction of the enzyme 11β-HSD1 - derived from 1, 2, 4-triazol by molecular modeling and precisely the molecular docking in solvated and unsolvated medium leads to the most stable complex. This study is based on the comparison of the theoretical and experimental results of the literature.
Results: Molecules 1,2,4 triazol derivatives interact differently with blood pressure enzyme (11β-HSD1) and confirm primary studies concerning1,2,4-triazol derivativesare good inhibitors for high blood pressure.
Conclusion: 11β-HSD1) enzyme with by molecular modeling by using molecular docking to confirm the experimental studies of 11β-HSD1 inhibitors reduce Medium
Keywords: Enzyme 11β-HSD1, molecular docking, solvation
Introduction
Hypertension is the most important risk factor associated with stroke and heart disease. We talk about high blood pressure when blood pressure is high in the arteries, so the heart makes more effort to pump blood into the blood vessels [1].
Theoretical methods, such as molecular modeling [2], now make it possible to study the interaction between enzymatic and biologically active molecules. Water plays a very important role in the biomolecular structure, so it is essential to represent the solvent around the solutes in the molecular modeling studies.
The explicit solvation method represents the solvent in a microscopic manner. In this case, a solvation cage is generated around the molecule. Quite simply, a number of solvent molecules (water) corresponding to its density are added randomly. A box or sphere of a defined size is created around the molecule that is filled with water molecules [3]. Many molecular docking software has been used in solvated media [4] such as Chemira, Hex, MolegroVertualDocker, and MOE (Molecular Operating Environment) software [4]. For our work we use the software MOE
Preparation of protein and ligands
1-Preparation of proteine
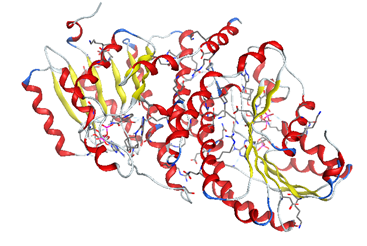
We downloaded our hypertension protein from the database "protein data bank (www.rcsb.org/pdb)" 11β-HSD1 (code: 4yyz) [5].
The enzyme was prepared for molecular docking by adding all the hydrogen atoms using standard procedures. The heteroatoms of co-crystallization were suppressed.
Minimization of the energy of the enzyme simplify equal to 3.928kcal / mol
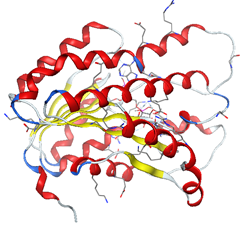
The preparation of the enzyme is done by creating the active site and isolating it to do the molecular docking in a well-designated active site.
2-Prepartion of ligands
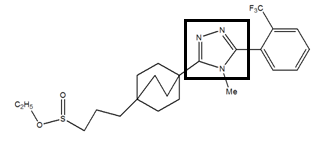
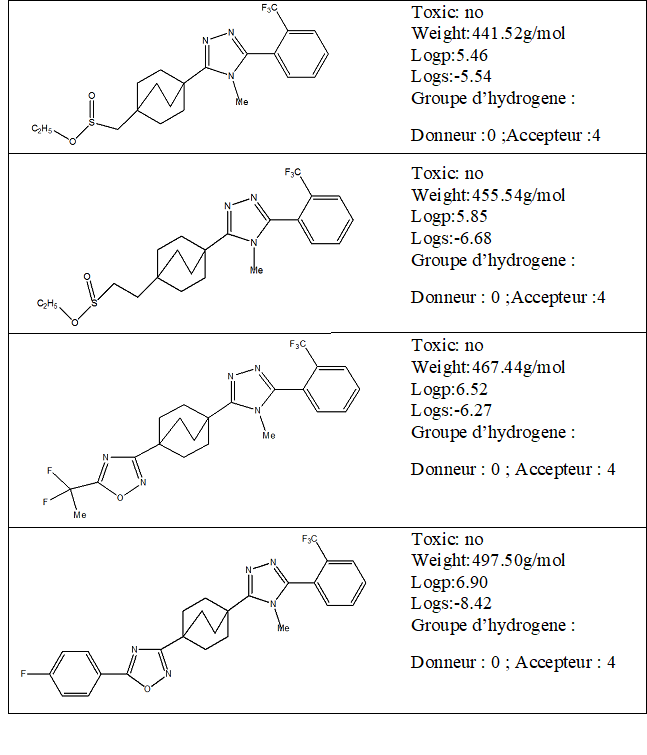
The 1, 2, 4-triazole molecule have been downloaded from the PubChemdatabase (www.pubchem.com). For the 1,2,4-triazole derivatives, the 1,2,4-triazole skeleton was retained and the side chain was modified.
Lipinski Rule
Before calculating the interactions between the enzyme and the four compounds, it is necessary to evaluate the parameters allowing their validation as a drug.
The Lipinski rule also known as the five rule (RO5) is a basic rule for evaluating or determining whether a chemical compound possesses pharmacological or biological activity. The rule was formulated by Christopher A. Lipinski in 1997; it is based on observation and informs us that the drugs administered orally are relatively smaller and moderatelylipophilic [6,7]. According to Table 1, the ligands satisfy the Lipinski rule. This additional information on other molecular properties confirms that these molecules are biologically active. The optimization of inhibitors was performed with hyperchem8.0.3software [8] to achieve the most stable conformation. In Table 2 we represent the energies of our molecules.
After the preparation of our enzyme and the four inhibitors, the examination of the enzymatic cavity and the calculation of the distances between the inhibitors and the side chains of the amino acids constituting the active site and the calculated energies is done using Of MOE molecular docking program.
The set of simulations that will be presented in this work was done using a solvent model explicitly represented. To study an explicit solvent molecule, it is necessary to solvate it, that is to say to immerse it entirely in a "solvent box". This method represents each molecule of water around solute as a given triatomic molecule, as shown schematically in Fig5.
A sphere (shape) of a defined dimension is created around the complex that is filled with water molecules (solvent).
The best placement of the ligand in the active site of the enzyme was chosen according to the best score.
The results of the interaction energies obtained by the MOE software are in the form ΔG (free energy), the latter being correlated with inhibitor constant Ki of the same complex according to the following formula [9-10]:
Constant of inhibition Ki is given in the following relation:
ΔG = -RT ln Ki
With R: constant gas (1.937cal .mol-1k-1),T: the temperature in kelvin (298k)
Ic50 values are determined experimentally in the literature [11].
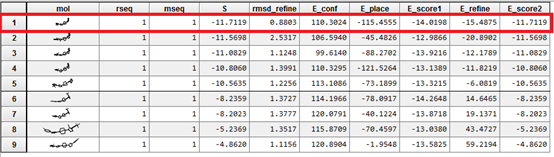
From the results obtained, it can be seen that for the 11β-HSD1_MOL4 complex at energy equal to -11.71 Kcal / Mol with a weak RMSD it is the most stable complex. It is concluded that MOL4 is probably the best inhibitor for the enzyme.
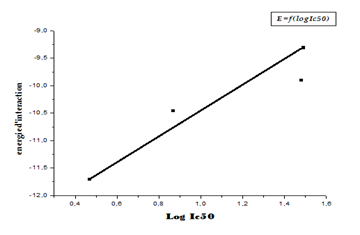
The linear regression analysis performed between the interaction energies and IC50 (log IC50) provides a scatter diagram with a correlation coefficient equal to 0.693. This value indicates that the two variables are highly correlated and thus demonstrate the high performance of the MOE program. From the results obtained, it can be seen that the 11β-HSD1_MOL4 complex with an energy equal to -11.71 Kcal / Mol with an RMSD equal to 0.88 (Å) is the most stable complex. It is concluded that L4 is probably the best inhibitor for the enzyme
The results of the molecular docking of the four complexes have an RMSD value of less than 2Å. The RMSD between 0Å and 2Åshe is considered low and between 2Å and 4Å is assumed to be average. RMSD greater than 4 Å is strong. The prediction of the ligand is acceptable if the value does not exceed 2 Å [12].
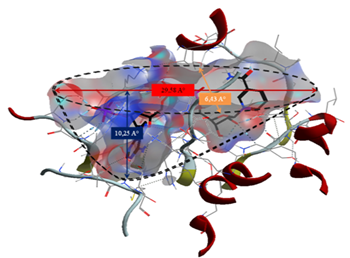
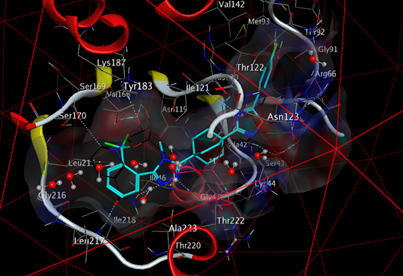
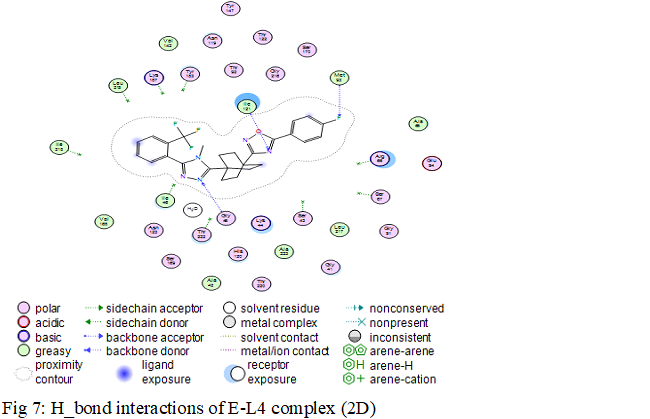
From the results obtained, the interactions formed between our ligands and the residues of the active site of the enzyme 11β-HSD1 are between 2.65 Å, and 4.31 Å.
Interactions between 2.5 Å and 3.1 Å are considered strong and those between 3.1 Å and 3.55 Å are assumed to be average. Interactions greater than 3.55 Å are weak or absent [13].
Following the results obtained, the ligand MOL4 forms strong interactions with the active site of the enzyme (11β-HSD1) and thus a better complementarity, it is concluded that the ligand MOL4 is probably the best inhibitor for enzyme (11β-HSD1)
The formation of a stable complex depends on the binding of the inhibitor to the active site. Fig 6 presented above shows that the ligand MOL4 forms in the enzymatic cavity formed by the residues of the active site, which means that there are interactions which stabilize the complex and then a better fixation of this inhibitor at the level of the cavity of the enzyme.
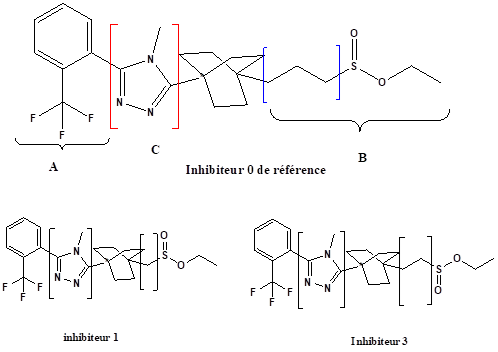
The results of molecular docking reveal that the energy of the complex formed by the inhibitor 2 is less than that of the complex formed by the inhibitor1; the complex inhibitor 2 is the most stable. It is concluded that the length of the chain (number of -CH2) increases the stability of the complex.
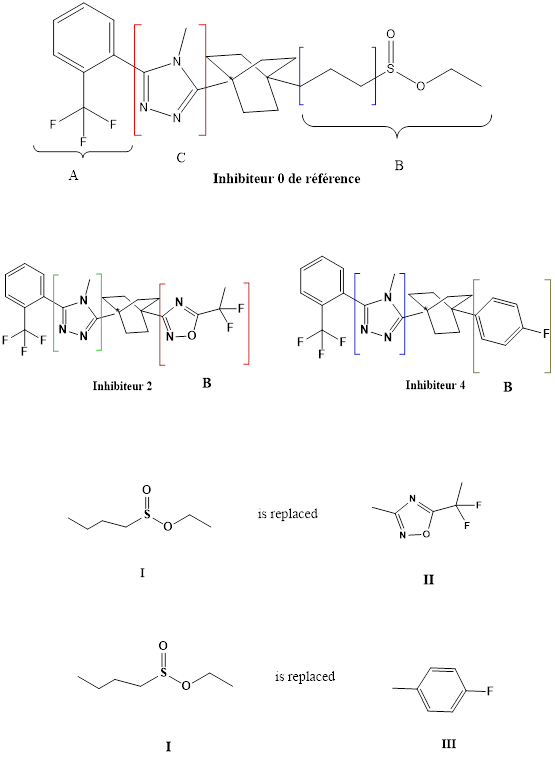
In the second step, the side chain B has been replaced by other groups:
- The substituent I by the substituent II,
- 2. The substituent I by the substituent III
According to molecular docking calculations, the complex formed with inhibitor 4 is more stable than the complex formed with inhibitor 2.
The fluorine-containing mesomer donor effect increases the conjugation of the aromatic ring as well as the stability of the complex formed between the enzyme 11β-HSD1 and inhibitor 4.From the results obtained, the complex inhibitor 4 is the most stable.
Conclusion
In order to determine the enzyme-substrate interactions we have carried out molecular docking calculations in order to find the most stable conformation which corresponds to the lowest energy adopted by the complex formed.
Water molecules in enzyme cavities can sometimes be a fundamental element. They are able to ensure the relay between the receptor and the ligand and thus create networks of hydrogen bonds.
The results of molecular docking show that the four compounds exhibit inhibitory activity, but the MO L4 inhibitor is the most stable because it has the lowest energy and the lowest IC50 inhibitory concentration according to the literature [11]. Therefore, theoretical calculations by molecular docking have confirmed the experimental results [11]. In conclusion the MOL4 inhibitor is probably the best inhibitor of our enzyme.
Images and Tables
|
molécules |
Energies (kcal/mol) |
|
Molécule01 |
-1851.97 |
|
Molécule02 |
-2133.77 |
|
Molécule03 |
-1933.89 |
|
Molécule04 |
-2560.45 |

|
Mode |
Droplet |
|
shape |
Sphere |
|
margin |
2.00 |
|
solvent |
Water |
|
delete |
far |
|
salt |
'' |
|
center |
0 |
|
|
|
|
align |
0 |
|
UpdatePotential |
1 |
|
ClashCutoff |
10.00 |
|
wallForce |
100.00 |
|
add_H |
1 |
|
verbose |
1 |
|
complexe |
Rmsd-refine(A°) |
Ic50(nM) |
ln Ic50 |
ki |
∆G(kcal/mol) |
|
11β-HSD1_MOL1 |
1.43 |
31.2 |
1.49 |
101.62.10-2 |
-9.31 |
|
11β-HSD1_MOL2 |
1.76 |
7.5 |
0.87 |
101.81.10-2 |
-10.46 |
|
11β-HSD1_MOL3 |
0.73 |
30.4 |
1.48 |
101.72.10-2 |
-9.90 |
|
11β-HSD1_MOL4 |
0.88 |
3.0 |
0.47 |
102.72.10-2 |
-11.71 |
|
molecule |
Ligand |
Receptor |
Interaction |
Distance (Å) |
E (kcal/mol) |
|
Mol1 |
O 45 |
NH2 ARG 66 |
(A) H-acceptor |
2.95 |
-7.8 |
|
Mol2 |
O 43 |
N ARG 66 |
(A) H-acceptor |
2.95 |
-7.8 |
|
O 43 |
NE ARG 66 |
(A) H-acceptor |
3.36 |
-1.2 |
|
|
O 43 |
NH2 ARG 66 |
(A) H-acceptor |
3.34 |
-1.2 |
|
|
5-ring |
N ILE 46 |
(A) pi-H |
4.31 |
-0.6 |
|
|
Mol3 |
F 28 |
NH2 ARG 66 |
(A) H-acceptor |
2.65 |
-1.0 |
|
Mol4 |
N 9 |
CA GLY 45 |
(A) H-acceptor |
3.30 |
-1.0 |
|
N 22 |
N ILE 121 |
(A) H-acceptor |
3.61 |
-0.6 |
|
|
F 32 |
N MET 93 |
(A) H-acceptor |
2.82 |
-1.0 |
References
NouhoumOuologuem? place de l'hypertension arterielle dans la pathologie cardiovasculaire dans le district de bamako en 2002 ? th?se de doctorat ; la Facult? de M?decine, de Pharmacie et d'Odontostomatologie ;universite de bamako, Mali2005.
R. Lavery. ? La mod?lisation mol?culaire : un outil pour ?tudier les macromol?cules biologiques et leurs interactions. De la mati?re au vivant : les syst?mes mol?culaires organis?s ?. P a r i s : CNRS- Images de la Recherche, p. 199?204, (1994).
Nathalie BASDEVANT ? Un Mod?le de Solvatation Semi-Implicite pour la Simulation des Macromol?cules Biologiques ? ; th?se de doctorat universite d'evryval-d'essonne, p56, 2003
Molecular operating environment (MOE) chemical computing group,montrealquebec , canada 10 ,2012
Robb, G.R., Boyd, S., Davies, C.D., Dossiter, A.G., Goldberg, F.W., Kemmitt, P.D., Scott, J.S., Swales, J.G." Design of pyrozolo-pyrimidines as 11beta-HSD1 inhibitors through optimisation of molecular electrostatic potential". Med chem. Comm 6: 926-934; 2016
View ArticleLipinski CA, Lombardo F, Dominy BW, Feeney PJ. "Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings". Adv. Drug Deliv. Rev. 46 (1-3): 3?26. (March 2001) 00129-0
View ArticleLipinski CA "Lead- and drug-like compounds: the rule-of-five revolution". Drug DiscoveryToday: Technologies 1 (4): 337?341. (December 2004). PMid:24981612
View Article PubMed/NCBIhyperchem (molecularmodeling system)hypercube ,inc.,1115NW 4th street, gainesville,FL32601,USA,2005.
Mar?chal, Yvus. "The hydrogen bond and the water molecule"Elsevier Science & Technology": eBook ISBN: 9780080469294. Hardcover ISBN:9780444519573, 2007.
HichamAyachi, ShirinJamshidi, MeriemMerad, YassineBendiabdallah, Said Ghalem,KhondakerMiraz Rahman "Design of New Inhibitors ofDipeptidyl Peptidase-4 in Type 2 Diabetes byComputer Simulations ?Der PharmaChemica, 9(2):17-29,2017
David R. Bauman a, Alan Whitehead b, Lisa C. Contino c, Jisong Cui a, Margarita Garcia-Calvo d, XinGub,Nancy Kevin b, Xiuying Mac, Lee-yuhPai c, Kashmira Shah c, XiaolanShen e, Sloan Stribling c, Hratch J. Zokian d, Joe Metzger c, Diane E. Shevell a, Sherman T. Waddell b,"Evaluation of selective inhibitors of 11b-HSD1 for the treatment of hypertension";Bioorganic & Medicinal Chemistry Letters 23 3650?3653,2013.
WL Jorgensen,"themanyrolesof computation in drug discovery", science-sciencemag.org.303:1813-1818, 2004.
A.Imberty, K. D. Hardman, J.P. Carver, S. P?rez:" Molecular of protein-carbohydrate interactions. Docking of monosaccharide's in the binding site ofconcanavaline a",glycobiology, 1, 631?642, 1991.