Mamadou Guy-Richard Koné
Email: guyrichardkone@gmail.com
© 2019 Sift Desk Journals. All Rights Reserved
VOLUME: 3 ISSUE: 1
Page No: 221-234
Mamadou Guy-Richard Koné
Email: guyrichardkone@gmail.com
Yafigui Traoré,*Mamadou Guy-Richard Koné, Ouanlo Ouattara, Nahossé Ziao
Laboratoire de Thermodynamique et de Physico-Chimie du Milieu, UFR SFA, Université Nangui Abrogoua, 02 BP 801 Abidjan 02, République de Côte-d’Ivoire
Mamadou Guy-Richard koné, QSAR APPROACH TO ESTIMATING THE ANALGESIC ACTIVITY OF A SERIES OF TRI-SUBSTITUTED PYRIMIDINE DERIVATIVES(2018)SDRP Journal of Computational Chemistry & Molecular Modelling 2(4) p:221-234
In the search for new molecules with improved analgesic activity, a series of twenty tri-substituted pyrimidine derivatives were subjected to a QSAR study. This study was conducted using Pearson Correlation Coefficients, Principal Component Analysis (PCA), Hierarchical Ascending Classification (HAC) and Multiple Linear Regression. The model obtained correlates the analgesic activity expressed by logAA, the energy of the highest occupied molecular orbital EHOMO , the dipole moment μD and the lipophilic coefficient ACD/logP. Statistical indicators R² = 91.55%; RMSE = 0.2592; F = 39.720; Q2LOO= 0.858 and R²p=0.765, establish the internal performance and stability of the model. While its external predictive ability has been proved by the statistical parameters Q2pred = 0.727 and RMSEP =0.403. The domain of applicability of the model developed, determined by the technique of levers and standardized residues, did not present any aberrant observations. The model thus established made it possible to determine the structures of eleven new tri-substituted pyrimidine derivatives having a better analgesic activity than the study molecules, at the dose of 10 mg/kg of body weight.
Keywords: Analgesic Activity, Quantum Chemistry, Pyrimidine Derivatives, QSAR Study
The International Association for the Study of Pain (IASP) defines pain as "an unpleasant sensation and an emotional experience in response to actual or potential tissue damage or described in these terms" [1]. It has several components including an affective and emotional component and a behavioral component. The pain changes the behavior. It can then result in the loss of jobs and even the rejection of the patient by relatives. Thus pain has a very high social cost and economic cost [2-4]. It has become a public health and ethical problem [5, 6], which mobilizes policy makers through care systems [7, 8]. The fight against pain is therefore necessary, obligatory and permanent. Several varieties of drugs exist for this purpose such as aspirin, paracetamol, codeine and morphine as well as many nonsteroidal anti-inflammatory drugs (NSAIDs). But many of these varieties have harmful side effects for the body [9]. In the case of NSAIDs, for example, the main adverse reactions commonly seen are nausea, vomiting, drowsiness, constipation, respiratory depression, gastrointestinal disturbances (mucosal irritation or perforation, ulcer, bleeding). and renal failure (renal failure) [10]. The design of new analgesics seeks to reduce side effects and enhance their effectiveness. This led to the synthesis of selective inhibitors of cyclooxygenase [11-13]. But research continues and every year, around the world, many molecules [14-16] and other natural substances are tested for their analgesic properties [17-20]. Pyrimidine derivatives have a good place in these studies. Indeed, many natural substances [21, 22] and synthetic products include this heterocycle with six vertices. They possess several interesting biological properties such as antimycobacterial [23], cardioprotective [24], antimalarial [25], antiparasitic [26] and, of course, analgesic and anti-inflammatory properties [27-31]. Pyrimidine derivatives are thus synthesized and analyzed for their analgesic properties around the world [12, 27-31]. In the classical method, researchers use the SAR methodology (Relation Structure Activity) [32, 33], in order to synthesize the molecules likely to have the best effect. The discovery of the analgesic activity of the compounds thus synthesized is done in vivo, using rats or mice, using a certain number of accepted techniques (tail-flick, hot-plate, writhing) [34] . This approach is expensive in synthetic products and time, so it is not without damage for the animals used. Alternative methods are then scanned. This is why the QSAR methodology, which has emerged for some decades [33, 35], and which links biological activity quantitatively to the molecular structure, has entered this research process [36-40]. It should be noted, however, that for the analgesic activities of pyrimidine derivatives, a very limited number use this methodology [31]. The aim of this work is to develop a QSAR model, by multiple linear regression, able to predict the analgesic activity of pyrimidine derivatives from physicochemical.
2.1 Analgesic Activity of Derivatives
The experimental database consists of a set of tri-substituted pyrimidine derivatives having central analgesic activity. These data come from studies by Vishal et al. [41] (9 derivatives) and Rajendra et al. [42] (11 derivatives) (Figure 1). The biological activity of these molecules was determined according to the same protocol, using the "Tail-Flick" technique [43, 44] which uses hot water at 55 ° C ± 0,5. The percentage inhibition (PI), pain (Table 1), were recorded at the time of 120 minutes after the oral intake of the drug candidate, by the animal (mouse), at the body doses D of 100 mg / kg [41] and 10mg / kg [42]. The inhibition percentages were converted to logAA, following expression 01, below (Table 1). This transformation makes the experimental values unbounded and takes into account both the D dose and the substance administered [45], through its molar mass M.
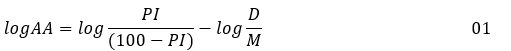
PI is expressed in%, D in mg per kg of body weight and M in g / mol.
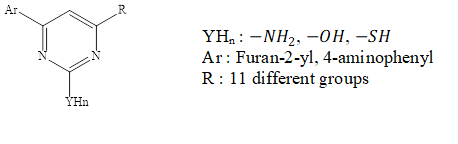
Figure 1: General structure of pyrimidine derivatives
2.2 Molecular Descriptors
The three descriptors of free-molecule selected for this study were calculated with the help of the Gaussian 03 [46] program, using the DFT and hybrid functional method B3LYP, used from the 6-31G (d,p) database and the ACD/ChemSketch program [47]. Gradient-corrected functionalities and hybrid functionalities such as B3LYP give better energies and are in good agreement with high-level ab initio methods [48]. The split-valence and double-dzeta bases (6-31G (d, p)) which is sufficiently extended and the fact of taking into account the polarization functions is important because it takes into account the free doublets of heteroatoms. These are the energy of the highest occupied molecular orbital (HOMO), the dipole moment and the octanol/water partition coefficient ACD/LogP. The energy of the HOMO is an energetic parameter of global molecular reactivity [49]. This energy expresses the susceptibility of the molecule to be attacked by electrophilic reagents. Because the highest occupied molecular orbital houses the last electrons of the molecule. Large values of (negative values) favor the electro-donating reactions of the molecule: this one easily releases its last electrons. Thus it has a higher nucleophilic power. The dipolar moment is related to the global distribution of the negative and positive charges of the molecule, it reflects the polarity of this one. Large values of the dipole moment mark the importance of the polar character of a molecule [50]. The octanol/water partition coefficient ACD/LogP is estimated according to Petrauskas et al. from atomic contributions and structural fragments taking into account intramolecular interactions [51]. This descriptor is a parameter of transport and passage through cell membranes. Large logP values are indicative of high solubility in lipids and good penetration into cell membranes but this implies low aqueous solubility. This can disadvantage the transport of the drug by the blood plasma. The descriptor values for the twenty derivatives are collated in Table 1.
2.3 Statistical Analyses
2.3 1 Data Analysis
Principal Component Analysis (PCA) [52] and Hierarchical Ascending Classification (HAC) [53, 54], implemented in XLSAT software [55], were employed.
PCA is a method of data analysis using projection. It is mainly applied to reduce the number of explanatory variables. It also produces a visualization of the observations in a space with two or three dimensions, in order to identify homogeneous groups or on the contrary atypical observations. It is for this purpose that this method is used in this work.
HAC is also a method of data analysis. It operates groupings into homogeneous classes of individuals in a set, using a similarity or dissimilarity measurement criterion and a grouping strategy based on an aggregation criterion. Successive groupings of individuals according to the aggregation criterion produce the dendrogram which is a hierarchy of partitions.
2.3.2 Construction and Estimation of the QSAR Model
Multiple Linear Regression (MLR) in the XLSTAT program is used to generate structural-activity analgesic models using the training set. This method establishes a linear relationship between logAA, expressing the biological activity, and representing the magnitude to be explained, and the three independent descriptors, which constitute the explanatory variables. The constructed models are evaluated by the values of the statistical indicators such as the coefficient of determination R², the standard error RMCE and the value of the test of Fisher F. These statistical indicators are defined by the expressions below [56, 57]:
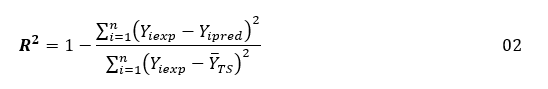
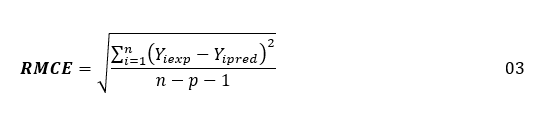
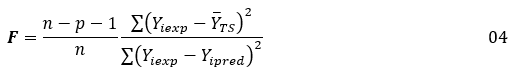
In these expressions: n is the number of molecules in the Training Set (TS), p is the number of descriptors in the model Yiexp and Yipred are the experimental and predicted values of biological activity for molecule YTS is its average value of biological activity for the training set.
2.3.3 Internal Prediction and Model Stability
To test the internal predictive character of the model developed, the leave-one-out (LOO) cross validation technique is used. This technique involves removing an observation from the formation set, then constructing a new model and finally predicting, by this model, the activity of the observation removed. The cross validation parameter Q2LOO according to the expression 05 below, is calculated at the end of the cycle.

The stability of the model is established by the randomization test of the dependent variable. To this end, by maintaining the matrix of independent descriptors, intact [58], the values of the analgesic activity are distributed, at random, to the compounds of the training set. After each distribution, a new model is constructed and its coefficient of determination R2YR is raised. Finally, the validation parameter of Roy et al., R2p is calculated according to expression 06 below [59, 60], after each randomization.

2.3.4 External Validation of the Model
As for the external predictive character of the model, it is established on the one hand by the prediction correlation coefficient and the predictive average prediction error (RMSEP), calculated according to the expressions 07 and 08 below:

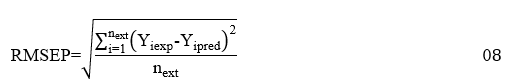
n_ext is the number of molecules of the test Yiexp and Yipred are the experimental and predicted values of the dependent variable for the molecule i belonging to the test set.

R2 Coefficient of correlation between predicted and observed activities with constant. R2 o Coefficient of correlation without constant, k slope of the correlation line of Ypred as a function of Yexp passing through the origin R'2 o Coefficient of correlation without constant, k' slope of the correlation line of Yexp as a function of Ypred passing through the origin. It is appropriate that these five criteria are satisfied [62] to validate a model.
3.1 Constitution of Training and Validation Sets
Below are some results of the data analysis:
Table 2: Pearson inter-correlation matrix (n) of the 3 selected descriptors and analgesic activity
|
Variables |
logAA |
ACD/LogP |
||
|
logAA |
1 |
|||
|
0.0703 |
1 |
|||
|
0.8514 |
-0.3091 |
1 |
||
|
ACD/LogP |
-0.1180 |
-0.3173 |
0.1580 |
1 |
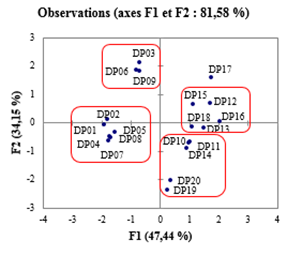
Figure 3: Representation of the 20 compounds in the factorial space (F1, F2) of the PCA, comprising 81.58% of the total information
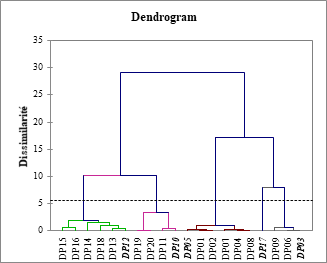
Figure 4: Dendrogram of the partition hierarchy of the 20 derivatives with truncation line at dissimilarity level 5,67 for 5 homogeneous classes
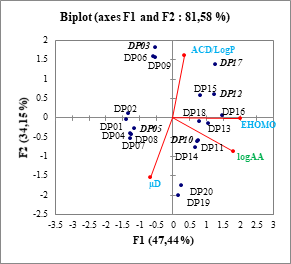
Figure 5: Circle of correlation of descriptors and magnitude explained
Table 2 and Figure 5 show the linear independence of the three descriptors and the strong correlation of the EHOMO descriptor with the biological activity.
The initial set of twenty compounds was subdivided into a training set (15) and a validation set (5), in the proportions 75% and 25% [63, 64], respectively using PCA and HAC. The PCA produced a visualization of the 20 observations in the factorial plane (F1, F2), representing 81.58% of the total information (figure 3). This visualization reveals four groups and an isolated observation which is DP17. As for the HAC, it has grouped into four homogeneous classes and an isolated observation all the observations, according to the dendrogram (Figure 4). To achieve this, Euclidean distances between observations, in the space defined by the descriptors, as dissimilarity criteria and Ward's method [65] as aggregation criteria were used. The four groups of the PCA as well as the four classes of the HAC contain the same observations. The five test-set derivatives (in bold italic) were selected by selecting one observation per group and the isolated observation DP17. The compounds of this set thus cover the whole of the dispersion and make it possible to take into account all the characteristics of the overall set [66].
3.2 Equation of the QSAR Model
Equation of the model (Equation 9) establishes a linear dependence between the analgesic activity expressed by logAA and the three descriptors μD , EHOMO and ACD/LogP. The positive sign of the coefficients of descriptors μD and EHOMO shows that the large values of these descriptors favor central analgesic activity. On the other hand, the negative sign of the coefficient of ACD/logP indicates that logAA and ACD/LogP evolve in opposite directions. High values of this third descriptor do not promote analgesic activity.
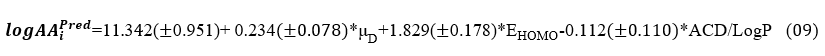
The value of the coefficient of determination R² indicates a model capable of explaining 91.55% of the experimental variance; the values estimated by the model are close to the experimental values. This result is corroborated by the relatively small value of the mean squared difference RMCE = 0.2592. As for the Fisher test, its F = 39.720 value confirms the significance of the model developed, in that it is greater than the required critical value F(11; 15; 0.05) = 2.507 [67]. Experimental and predicted values and residues are summarized in Table 1.
According to the absolute values of the normalized coefficients (Figure 6), the importance of the weight of the descriptors involved in the model (Equation 9) decreases in the following order: . The HOMO energy has the largest contribution with 0.9767 and the smallest EHOMO > μD > ACD/logP with (-0.1046), while μD has 0.3253 as the contribution. The analgesic activity of these derivatives is therefore favored by molecules having a high nucleophilic character, a high polarity and low values of the lipophilic coefficient. Any proposal for a molecule to improve the central analgesic activity of these derivatives should take into account these criteria.
3.3 Internal and External Validations
The leave-one-out (LOO) cross-validation and randomization techniques of the explained variable were applied on the fifteen compounds of the training set. The statistical parameters of these validations are: Q2LOO =0.8576 and R2p=0.7646, after 10 randomizations [68]. The cross-correlation coefficient Q2LOO > 0.5 which is the minimum required value, according to Tropsha et al. [69]. The model developed therefore has a satisfactory internal predictive character. Also, the average value of the Roy et al. Parameter R2p , obtained after the ten randomizations is also greater than the minimum required value which is also 0.5 as well. This shows that the constructed model is not the result of chance [59-66]. All of these statistical parameters indicate that the developed model has a satisfactory internal predictive character and can be considered robust [56] and stable.
According to the external prediction parameters, the elaborated model is able to explain the activity of the unknown compounds of the test set with a prediction correlation coefficient Q2pred = 0.7271 and a Predictive Mean Prediction (RMSEP) error of 0.4033. This value is less than the standard deviation of the experimental values of the formation set which is σ = 0.7905.
Concerning the validation criteria of Tropsha and Golbraikh, we have:
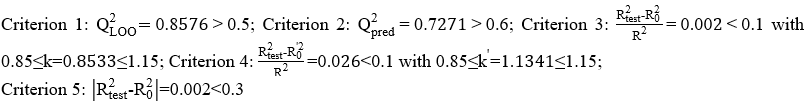
These different values make it possible to affirm that the model developed enjoys a good external predictive performance
3.4 Applicability Domain
The applicability domain of the model was defined using the standardized residue technique represented as a function of the levers hii [70], calculated according to the expression 10 below.

Where xi is the line vector of the descriptors of the compound i and X is the matrix built on the values of the descriptors of the model and the compounds of the training set. The exponent t refers to the matrix or transposed vector. The MINITAB software was used to calculate the levers of the molecules of the training set as well as those of the molecules of the test set. Figure 7 below shows the graph obtained. For the N = 15 compounds of the formation set and the k = 3 descriptors of the model, the threshold value of the levers h*= 3 k+1/N is 0.800. The standard deviation of the experimental values of the training set being σ = 0.7905. The extreme values of the residues are (± 3σ), ie -2.372 and +2.372. These different values delimit the domain of applicability of the model [70-73].
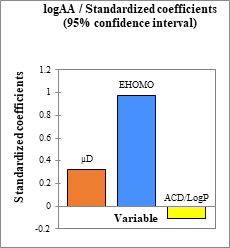
Figure 6: Standardized coefficients of descriptors of the developed model
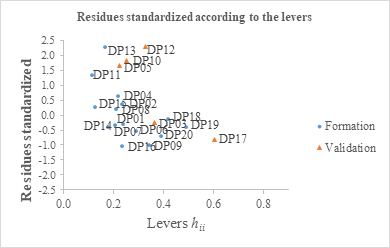
Figure 7: William's plot of standardized residues according to the leverages
The graph (Figure 7) shows that all the observations of the training and validation sets belong to the defined applicability domain. No aberrant observations can be reported for the model developed [72]. Thus, for the model developed to predict the analgesic activity of a new molecule, its lever must be below the threshold value h* = 0.800. This molecule must belong to the domain of applicability of the model [74].
The experimental and predicted values as well as the residues obtained for the compounds of the two sets are given in Table 1. Figures 8 and 9 show the regression line of the predicted and experimental values as well as the graph of similarity.
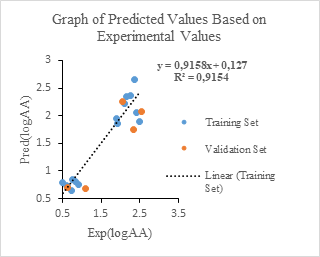
Figure 8: Graph of experimental values versus logAA predicted values
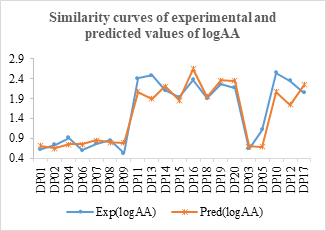
Figure 9: Similarity curves of the experimental values and predicted by the model
The two point clouds are in adequacy for the two sets according to figure 8. The similarity curves of figure 9 show a slight overestimation of the predicted values with respect to the experimental values.
Table 1: Designation codes, Natures of substituents and percentages of pain inhibition after 120 minutes of derivation of the derivative, Values of the three model descriptors, experimental and predicted values and calculated residues for each compound of the training and validation set.
|
Training Set |
|||||||||||||
|
Compounds |
Ar |
R |
YHn |
(D) |
(eV) |
ACD/LogP |
PI(%) |
Exp(logAA) |
Pred(logAA) |
Residual |
|||
|
DP01 |
C4H3O |
BrC6H4 |
OH |
4.252 |
-6.208 |
2.440 |
57.03 |
0.623 |
0.713 |
-0.090 |
|||
|
DP02 |
C4H3O |
BrC6H4 |
OH |
4.278 |
-6.227 |
2.790 |
61.97 |
0.733 |
0.645 |
0.088 |
|||
|
DP04 |
C4H3O |
ClC6H4 |
SH |
4.321 |
-6.216 |
2.100 |
74.90 |
0.909 |
0.751 |
0.158 |
|||
|
DP06 |
C4H3O |
ClC6H4 |
SH |
2.430 |
-5.885 |
3.640 |
59.69 |
0.604 |
0.744 |
-0.141 |
|||
|
DP07 |
C4H3O |
FC6H4 |
NH2 |
3.928 |
-6.154 |
1.470 |
69.20 |
0.760 |
0.844 |
-0.084 |
|||
|
DP08 |
C4H3O |
FC6H4 |
NH2 |
4.092 |
-6.175 |
1.820 |
72.24 |
0.850 |
0.805 |
0.045 |
|||
|
DP09 |
C4H3O |
FC6H4 |
NH2 |
1.791 |
-5.820 |
3.010 |
56.27 |
0.516 |
0.783 |
-0.267 |
|||
|
DP11 |
C6H6N |
FC6H4 |
NH2 |
4.018 |
-5.428 |
2.610 |
90.23 |
2.413 |
2.065 |
0.347 |
|||
|
DP13 |
C6H6N |
C8H10O2 |
NH2 |
2.554 |
-5.358 |
2.160 |
90.45 |
2.484 |
1.901 |
0.583 |
|||
|
DP14 |
C6H6N |
C9H13O3 |
NH2 |
3.554 |
-5.342 |
1.710 |
78.46 |
2.108 |
2.214 |
-0.106 |
|||
|
DP15 |
C6H6N |
C7H7 |
NH2 |
2.739 |
-5.346 |
3.130 |
75.37 |
1.927 |
1.859 |
0.068 |
|||
|
DP16 |
C6H6N |
C8H10N |
NH2 |
2.982 |
-4.966 |
2.780 |
88.63 |
2.376 |
2.649 |
-0.272 |
|||
|
DP18 |
C6H6N |
2-C5H4N |
NH2 |
1.958 |
-5.310 |
1.330 |
75.41 |
1.907 |
1.942 |
-0.035 |
|||
|
DP19 |
C6H6N |
4-C5H4N |
NH2 |
5.441 |
-5.529 |
1.290 |
87.22 |
2.254 |
2.361 |
-0.107 |
|||
|
DP20 |
C6H6N |
3-C5H4N |
NH2 |
5.040 |
-5.478 |
1.360 |
84.79 |
2.166 |
2.353 |
-0.186 |
|||
|
Validation Set |
|||||||||||||
|
DP03 |
C4H3O |
BrC6H4 |
OH |
2.342 |
-5.875 |
3.980 |
57.79 |
0.635 |
0.703 |
-0.068 |
|||
|
DP05 |
C4H3O |
ClC6H4 |
SH |
4.329 |
-6.234 |
2.450 |
81.74 |
1.110 |
0.682 |
0.428 |
|||
|
DP10 |
C6H6N |
ClC6H4 |
NH2 |
4.716 |
-5.473 |
3.240 |
92.24 |
2.546 |
2.076 |
0.471 |
|||
|
DP12 |
C6H6N |
C7H6O |
NH2 |
1.597 |
-5.302 |
2.490 |
88.31 |
2.344 |
1.744 |
0.600 |
|||
|
DP17 |
C6H6N |
C14H9 |
NH2 |
3.433 |
-5.097 |
5.140 |
75.26 |
2.042 |
2.252 |
-0.210 |
|||
The absolute values of the residues for both sets are all smaller than the standard deviation of the training set values σ = 0.7905
3.5 Improvement of Analgesic Activity
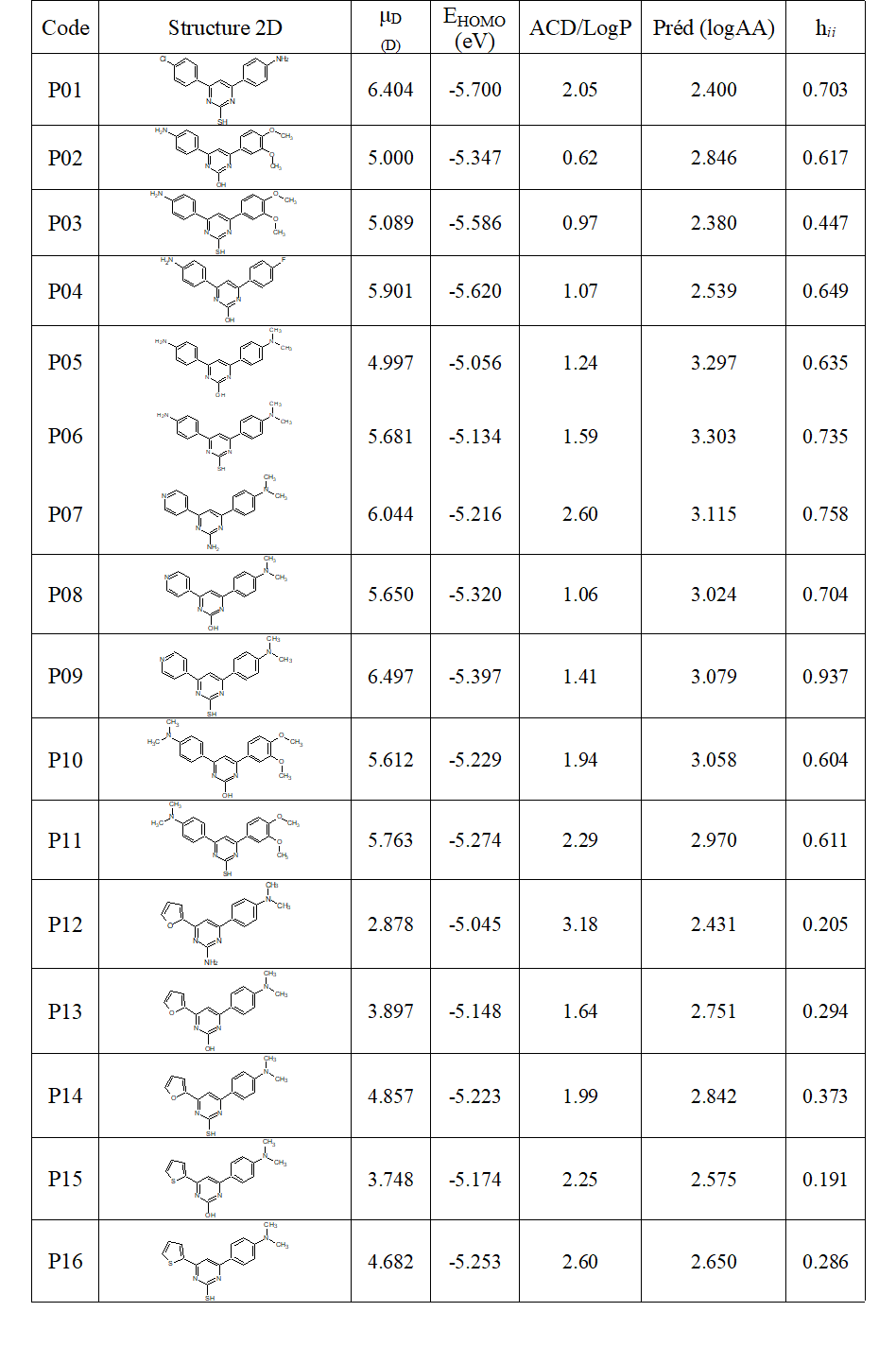
In order to obtain derivatives having an analgesic activity higher than the maximum experimental activity, structural modifications were made to the molecules having the best experimental activities, namely: DP10 with 2.546; DP13 with 2.484; DP11 with 2.413 and DP16 with 2.376. Sixteen (16) tri-substituted derivatives were then proposed. After optimization, then calculation of the frequencies at the level B3LYP/6-31G (d, p), the dipole moment as well as the energy of the HOMO of each of the sixteen proposed molecules were recorded. The coefficient of lipophilicity ACD/logP was calculated for each of the molecules. Equation 9 was used to calculate logAA for each proposition. Their levers were also calculated. Table 3 contains the designation codes, the 2D structures, the values of the three descriptors, those of logAA predicted as well as the levers of the proposed molecules.
Of the 16 proposals, 12 have a logAA value greater than the maximum experimental value. A proposition has a leverage greater than the threshold, of value 0.937. This is P09 which is part of the twelve. This outlier could be due to the high value of the dipole moment, ie 6.497, which is the highest. Thus the model was able to find the structures of eleven new molecules similar to the basic molecules used and having a higher central analgesic activity. It is P02, P05, P06, P07, P08, P10, P11, P13, P14, P15 and P16.
Table 3: Designation codes, 2D structures, values of the three descriptors and logAA predicted by the model (Equation 9) as well as the hii levers of the proposed molecules
This work has shown that it is possible to explain the central analgesic activity of tri-substituted pyrimidine derivatives by the , and descriptors. Both quantum descriptors were calculated by the DFT method at the B3LYP/6-31 (d, p) theory level. Multiple linear regression (MLR) has been used to develop a QSAR model. The energy of the highest occupied molecular orbital has, in this model, the largest contribution followed by the dipolar momentum μD and finally the coefficient of lipophilia ACD/LogP. The established model can be considered reliable, robust and efficient capable of predicting the analgesic activity of new pyrimidine compounds belonging to the defined field of applicability, given the values of the different statistical estimators (R2 = 91.55%, RMCE = 0.2592, F = 39.7201, Q2LOO= 85.76, Q2pred = 72.71% ). Thus, this model made it possible to determine the structures of eleven new tri-substituted derivatives of pyrimidine having a better analgesic activity than that of the starting molecules. A synthesis of these molecules should be performed to verify these theoretical predictions.
B. Calvino, Neuromodulation-neurostimulation : physiopathologie de la douleur et cibles neurochirurgicales, Douleurs Évaluation - Diagnostic - Traitement (2011), 12, 224-233
F. T. Luethi, Quels sont les Obstacles à la Gestion de la Douleur perçus par les Infirmières ? Université De Lausanne Juillet (2013)
H. Breivik, B.y Collett, V. Ventafridda, R. Cohen, D. Gallacher, Survey of Chronic Pain in Europe : Prevalence, Impact On Daily Life, And Treatment, European Journal of Pain, (2006), 10, 287-333, PMid:16095934
View Article PubMed/NCBIF. Atallah, Y. Guillermou, L'homme et sa douleur : dimension anthropologique et sociale, Annales Françaises d'Anesthésie et de Réanimation, (2004), 23, 722-729. PMid:15324961
View Article PubMed/NCBIV. Tomlinson, The Development of an Opioid PGD To Improve Severe Pain Management in A&E Departments, Manchester Metropolitan University (2011).
Direction de l'Hospitalisation et de l'Organisation des Soins, Ministère de la Santé, de la Famille et des Personnes Handicapées (France), Lutter Contre la Douleur, Guide d'Orientation, Organiser la Lutte Contre la Douleur dans les Etablissements de Santé, 0AA68d01
C. Cousin (Doctorant), Du droit du patient de recevoir des soins antalgiques à l'obligation du médecin de prendre en charge la douleur, Médecine & Droit (2012) 158-160.
View ArticleN. Lelièvre, Douleur et droit : quelles évolutions en dix ans ? Douleurs Évaluation - Diagnostic - Traitement (2009), 10, 29-34.
C. Capet, C. Bentot, L. Druesne, Ph. Chassagne, J. Doucet, Les effets indésirables des anti-inflammatoires non stéroïdiens (AINS) chez le sujet âgé, La Revue de Gériatrie, Tome 26, N°5 Mai (2001).
Centre Belge D'Information Pharmaco-thérapeutique, Répertoire Commenté Des Médicaments (2013), 229-247 www.cbip.be (08/08/2015)
J.-Y. Jouzeau, M. Daouphars, A. Benani, P. Netter, Pharmacologie et classification des inhibiteurs de la cyclooxygenase, Gastroenterol Clin Biol (2004);28:C7-C17. 95274-8
View ArticleA. M. Alanazi, A. S. El-Azab, I. A. Al-Suwaidan, K. Eldin H. El Tahir, Y. A. Asiri, N. I. Abdel-Aziz, A. A.-M. Abdel-Aziz, Structure-based design of phthalimide derivatives as potential cyclooxygenase-2 (COX-2) inhibitors: Anti-inflammatory and analgesic activities, European Journal of Medicinal Chemistry, (2015), 92, 115-123 PMid:25549551
View Article PubMed/NCBID. Mishra, G. Ghosh, P. S. Kumar And P. K. Panda, An Experimental Study of Analgesic Activity of Selective COX-2 Inhibitor With Conventional NSAIDs, Asian Journal of Pharmaceutical and Clinical Research (2011), 4(1), 78-81.
F. A.-F. Ragab, N. M. Abdel-Gawad, H. H. Georgey, and M. F. Said, Pyrazolone Derivatives: Synthesis, Anti-inflammatory, Analgesic, Quantitative Structure–Activity Relationship and in Vitro Studies, Chem. Pharm. Bull. (2013), 61(8), 834-845
View ArticleM. M. Suleiman, S. G. Isaev, O.V. Klenina and V. V. Ogurtsov, Synthesis, biological activity evaluation and QSAR studies of novel 3-(aminooxalyl-amino)-and 3-(carbamoyl-propionylamino)-2phenylamino-benzoic acid derivatives, J. Chem. Pharm. Res., (2014), 6(5),1219-1235.
P. Datar, QSAR and Synthesis of a Novel Biphenyl Carboxamide Analogue for Analgesic Activity. Mod Chem appl., (2015), 3: 148.
R. Jayaraman, T. Anitha, V. D. Joshi, Analgesic And Anticonvulsant Effects Of Acorus Calamus Roots In Mice, Int. J. PharmTech Res.(2010),2 (1), 552-555.
R. R. Chatter, S. Tarhouni, R. Kharrat, Criblage de l'effet anti-inflammatoire et analgésique des algues marines de la mer méditerranée. Les Archives de l'Institut Pasteur de Tunis, (2012), (1-4), 19-28.
N. Ouédraogo, M. Lompo, RW. Sawadogo, A. Tibiri, A.-E. Hay, J. Koudou, M.-G. Dijoux, I.P. Guissou, Étude des activités anti-inflammatoire, analgésique et antipyrétique des décoctés aqueux des feuilles et des racines de Pterocarpus erinaceus Poir. (Fabaceae).
K. Arab, O. Bouchen A K et K. Yahiaoui, Évaluation de l'activité biologique des feuilles de l'olivier sauvage et cultivé, Afrique SCIENCE, (2013), 09(3) 159 - 166.
I. M. Lagoja, Pyrimidine as Constituent of Natural Biologically Active Compounds Chemistry and Biodiversity, (2007), 2(1), 1-50.
K. S. Jain1, T. S. Chitre, P. B. Miniyar, M. K. Kathiravan, V. S. Bendre, V. S. Veer, S. R. Shahane et C. Biological and medicinal significance of pyrimidines, J. Shishoo, Current Science, (2006), 90(6), 793-803.
A. R. Trivedi, D. K. Dodiya, N. R. Ravat, et V. H. Shah, Synthesis and biological evaluation of some new pyrimidines via a novel chalcone series, Arkivoc (2008), 131-141.
M. S. Mohamed, S. M. Awad and A. I. Sayed, Molecules (2010), 15, 1882-1890. PMid:20336018 PMCid:PMC6257238
View Article PubMed/NCBIA. Agarwal, K. Srivastava, S. K. Puri, and Chauhan, Synthesis of 2,4,6-trisubstituted pyrimidines as anti-malarial agents, P. M. S. Bioorg. med. chem. (2005), 13, 4645-4650. PMid:15896965
View Article PubMed/NCBIL. Sun, J. Wu, L. Zhang, M. Luo and D. Sun, Synthesis and Antifungal Activities of Some Novel Molecules, (2011), 16: 5618-5628.
J. K. Gupta, P. K. Sharma, R. Dudhe, S. C. Mondal, A. Chaudhary And K. Prabhakar, Synthesis And Analgesic Activity of Novel Pyrimidine Derivatives of Coumarin Moiety, Verma, Acta Poloniae Pharmaceutica - Drug Research, (2011), 68(5), 785-793.
S. G. Khanage, A. Raju, P. B. Mohite, R. B. Pandhare, Analgesic Activity of Some 1,2,4-Triazole Heterocycles Clubbed with Pyrazole, Tetrazole, Isoxazole and Pyrimidine. Advanced Pharmaceutical Bulletin, (2013), 3(1), 13-18,
H. N. Hafez, O. K. Al-Duaij, A.-R. Barakat A. El-Gazzar, Design, Synthesis and Pharmacological Evaluation of New Nonsteroidal Anti-Inflammatory Derived from 3-Aminobenzothieno[2,3-d]pyrimidines, International Journal of Organic Chemistry, (2013), 3, 110-118.
View ArticleA. A. Abu-Hashem, and M. M. Youssef, Synthesis of New Visnagen and Khellin Furochromone Pyrimidine Derivatives and Their Anti-Inflammatory and Analgesic Activity, Molecules. (2011), 16, 1956-1972. PMid:21358587 PMCid:PMC6259619
View Article PubMed/NCBIM. M. Hanna, New pyrimido[5,4-e]pyrrolo[1,2-c]pyrimidines : Synthesis, 2D-QSAR, anti-inflammatory, analgesic and ulcerogenicity studies, European Journal of Medicinal Chemistry (2012), 55, 12-22. PMid:22818041
View Article PubMed/NCBIM. Haghdadi, F. S. Kenary, H. G. H. Basra, Structure-activity-relationships study of 2-thienyl-4-furyl-6-aryl pyridine skeleton as anti-cancer drugs by DFT method, Organic Chemistry, An Indian Journal Vol. (2014), 10 (9), 371-376.
H. V. De Waterbeemd and S. Rose, Quantitative Approaches to Structure-Activity Relationships, Wermuth's The Practice of Medicinal Chemistry (Elsevier Ltd) (2008): Chapter 23, 491-513,
P. Milind and Y. Monu, Laboratory Models for Screening Analgesics, Int. Research Journal of Pharmacy, (2013), 4 (1), 15-19
M. Shahlaei, Descriptor Selection Methods in Quantitative Structure-Activity Relationship Studies: A Review Study, American Chemical Society (2013), 113 (10), 8093-8103
J. S. N'dri, M. G-R. Koné, C. G. Kodjo, S. T. Affi A. L. C. Kablan, Z. A. Ouattara and N. Ziao, Quantitative Structure antifungal Activity Relationship (QSAR) study of a series of Schiff bases derivatives from4aminobenzenesulphonamide By DFT method, IOSR: Journal Of Pharmacy, (2017), 7(4): 27-33.
K. N. N'guessan, M. G-R. Koné, K. Bamba, W. P. Ouattara, N. Ziao, Quantitative Structure Anti-Cancer Activity Relationship (QSAR) of a Series of Ruthenium Complex Azopyridine by the Density Functional Theory (DFT) Method, Computational Molecular Bioscience, (2017), 7: 19-31
View ArticleY. H. Kpidi, O. B. Yapo, M. G-R. Koné, G. A. Gadji, A. E. J. E. Gnagne, J. S. N'dri, and N. Ziao, Monitoring and Modeling of Chlorophyll-a Dynamics in a Eutrophic Lake: M'koa Lake (Jacqueville, Ivory Coast), American Journal of Environmental Protection, (2018), 6(1): 1-9.
View ArticleJ. S. N'dri, M. G-R. Koné, C. G. Kodjo, A. L. C. Kablan, L. Ouattara, O. Ouattara and N. Ziao, Combining of DFT and QSAR Results to Predict the Antibacterial Activity of a Series of Azetidinones derived from Dapsone as Inhibitors of Bacillus Sub-tilis and Pseudomonas Aeruginosa, Journal of Computational Chemistry & Molecular Modelling, (2018), 2(2): 1-9.
View ArticleD. Soro, L. Ekou, M. G-R. Koné, T. Ekou, S. T. Affi, L. Ouattara and N. Ziao, Prediction of the Inhibitory Concentration of Hydroxamic Acids by DFT-QSAR Models on Histone Deacetylase 1, International Research Journal of Pure & Applied Chemistry, (2018), 16(2): 1-13,
View ArticleV. D. Joshi, M. D. Kshirsagar and S. Singhal, Synthesis and Pharmacological Study of Some Novel Pyrimidines, Der Pharmacia Sinica, (2012), 3 (3), 343-348.
R. P. Yejella and S. R. Atla, A Study of Anti-inflammatory and Analgesic Activity of New 2,4,6-Trisubstituted Pyrimidines, Chem. Pharm. Bull. (2011), 59(9), 1079-1082.
View ArticleK. - Siransy G., I. Nguessan G., Dally I., Mohou B., Kamenan A., Kouakou L., Kablan B. J., Etude De L'activité Analgésique De L'extrait Méthanolique Des Feuilles De Gossypium Hirsutumlinn. (Malvaceae), J. Sci. Pharm. Biol., (2010), 11(1), 6-12.
D. Mishra, G. Ghosh, P. S. Kumar and P. K. Panda, Experimental Study of Analgesic Activity Of Selective COX-2 Inhibitor With Conventional NSAIDs, Asian J. of Pharmaceutical and Clinical Research. (2011), 4(1), 78-81
J. K Malik, H. Soni, Singhai A K, and H. Pandey, QSAR - Application in Drug Design, Int. J. Pharmaceutical Research & Allied Sciences. (2013), 2(1), 1-13.
Gaussian 03, Revision C.01, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, Gaussian, Inc., Wallingford CT, (2004).
ACD/LogP, v.10, Advanced Chemistry Development, Inc., Toronto, On, Canada, (2007).
B. G. Johnson, P. M. Gill, J. A. Pople, The performance of a family of density functional methods, The Journal of Chemical Physics, (1993), 98, 5612-5626.,
View ArticleS. Chtita, M. Larif, M. Ghamali, M. Bouachrine, T. Lakhlifi, DFT-based QSAR Studies of MK801 derivatives for non competitive antagonists of NMDA using electronic and topological descriptors, Journal of Taibah University for Science (2014) .
M. W. Wong, M. J. Frisch, K. B. Wiberg, «Solvent effects: The mediation of electrostatic effects by solvents» J. Am. Chem. Soc., (1991), 113(113), 4776-4782,
View ArticleR. Mannhold, G. I. Poda, C. Ostermann, I. V. Tetko, Calculation of Molecular Lipophilicity: State-of-the-Art and Comparison of LogP Methods on More Than 96,000 Compounds, J. of Pharmaceutical Sciences, (2009), 98(3), 861-893. PMid:18683876
View Article PubMed/NCBIS. Vahdani and Z. Bayat, A Quantitative Structure-Activity Relationship (QSAR) Study of Anti-cancer Drugs, Der Chemica Sinica, (2011), 2 (4):235-243.
Amadou H., Laouali M. S., Manzola A., Analyses Physico-Chimiques et Bactériologiques des Eaux de Trois Aquifères de la Région de Tillabéry : Application des Méthodes d'analyses Statistiques Multi Variées, Larhyss Journal, (2014), 20, 25-41.
R. Kiralj and M. M. C. Ferreira, Basic Validation Procedures for Regression Models in QSAR and QSPR Studies: Theory and Application, J. Braz. Chem. Soc., (2009), 20(4), 770-787.
View ArticleS. Chtita, M. Ghamali, R. Hmamouchi, B. Elidrissi, M. Bourass, M. Larif, M. Bouachrine and T. Lakhlifi, Investigation of Antileishmanial Activities of Acridines Derivatives against Promastigotes and Amastigotes Form of Parasites Using QSAR Analysis, Advances in Physical Chemistry, (2016),1-16.
View ArticleR. Veerasamy, H. Rajak, A. Jain, S. Sivadasan, C. P. Varghese and R. K Agrawal, Validation of QSAR Models - Strategies and Importance, Inter. J. of Drug Design and Discovery,(2011), 2(3), 511-519
O. Ouattara, T. S. Affi, M. G.-R. Kone, K. Bamba, N. Ziao, Can Empirical Descriptors Reliably Predict Molecular Lipophilicity ? A QSPR Study Investigation, Int. J. of Engineering Research and Application, (2017), 7(5), 50-56.
View ArticleC Rücker, G. Rücker, and M. Meringer, y-Randomization and Its Variants in QSPR/QSAR. J. Chem. Inf. Model. (2007), 47, 2345-2357. PMid:17880194
View Article PubMed/NCBIV. Vlaia, T. Olariu, L Vlaia, M. Butur, C. Ciubotariu, M. Medeleanu and D. Ciubotariu, Quantitative Structure-Activity Relationship (Qsar). Iv. Analysis Of The Toxicity Of Aliphatic Esters By Means Of Weighted Holistic Invariant Molecular (Whim) Descriptors. Farmacia, (2009), 57(4), 511-522
W. Li, F. Xiao, M. Zhou, X. Jiang, J. Liu, H. Si, M. Xie, X. Ma, Y. Duan, H. Zhai, 3D-QSAR study and design of 4-hydroxyamino α pyranone carboxamide analogues as potential anti-HCV agents, Chemical Physics Letters (2016), 661, 36-41,
View ArticleA. Golbraikh and A. Tropsha, Predictive QSAR modeling based on diversity sampling of experimental datasets for the training and test set selection, Molecular Diversity, (2002),5: 231-243, PMid:12549674
View Article PubMed/NCBIL. Qin, X. Zhang, Y. Chen, L. Mo, I D, H. Zeng and Y. Liang, Predictive QSAR Models for the Toxicity of Disinfection Byproducts. Molecules (2017), 9; 22(10). pii: E1671.
T. M. Martin, P. Harten, D. M. Young, E. N. Muratov, A. Golbraikh, H. Zhu and A. Tropsha, Does Rational Selection of Training and Test Sets Improve the Outcome of QSAR Modeling? J. Chem. Inf. Model. (2012), 52, 2570-2578. PMid:23030316
View Article PubMed/NCBIZ. Hajimahdi, F. Safizadeh and A. Zarghi., QSAR Analysis for Some 1, 2-Benzisothiazol-3-one Derivatives as Caspase-3 Inhibitors by Stepwise MLR Method, Iranian Journal of Pharmaceutical Research (2016), 15 (2): 439-448. PMid:27642314 PMCid:PMC5018271
PubMed/NCBIA. Labenne, M. Chavent, V. Kuentz-Simonet et J. Saracco, Classification Ascendant Hiérarchique avec Contrainte de Proximité Géographique.
K. Roy et al., A Primer on QSAR/QSPR Modeling, Chapter 2 Statistical Methods in QSAR/QSPR, Springer Briefs in Molecular Science, (2015), 37-59.
View ArticleA. Fortuné. Techniques de Modélisation Moléculaire appliquées à l'Etude et à l'Optimisation de Molécules Immunogènes et de Modulateurs de la Chimiorésistance, Médicaments. Université Joseph-Fourier - Grenoble I, 2006. Français.
R. Kiralj and M. M. C. Ferreira. Basic Validation Procedures for Regression Models in QSAR and QSPR Studies: Theory and Application. J. Braz. Chem. Soc., (2009), 20(4), 770-787.
View ArticleJ. Shamsara, Ezqsar: An R Package for Developing QSAR Models Directly From Structures. The Open Medicinal Chemistry Journal, (2017), 11, 212-221. PMid:29387275 PMCid:PMC5748834
View Article PubMed/NCBIJ. Jaworska, N. Nikolova-Jeliazkova and T. Aldenberg, QSAR Applicability Domain Estimation by Projection of the Training Set in Descriptor Space: A Review, ATLA 33, (2005).445–459.
M. Ghamali, S. Chtita, M. Bouachrine and T. Lakhlifi, Méthodologie générale d'une étude RQSA/RQSP, Revue Interdisciplinaire, 2016, 1(1), 1-6
S. Chtita, M. Ghamali, M. Larif, R. Hmamouchi, M. Bouachrine and T. Lakhlifi, Quantitative structure–activity relationship studies of anticancer activity for Isatin (1H-indole-2,3-dione) derivatives based on density functional theory, Int. J. Quantitative Structure-Property Relationships (2017), 2 (2), 90-115.
View ArticleD. Gadaleta, G. F. Mangiatordi, M. Catto, A. Carotti and O. Nicolotti, Applicability Domain for QSAR Models: Where Theory Meets Reality, Int. J. Quantitative Structure-Property Relationships, (2016), 1 (1), 45-63.
View ArticleS. Chtita, M. Larif, M. Ghamali, M. Bouachrine, T. Lakhlifi, DFT-based QSAR Studies of MK801 derivatives for non competitive antagonists of NMDA using electronic and topological descriptors, J. of Taibah University for Science. (2014), 9 (2), 143-154.
View Article