J. Thulasidhasan
Email: drthulasi86@gmail.com
© 2019 Sift Desk Journals. All Rights Reserved
VOLUME: 5 ISSUE: 1
Page No: 539-542
J. Thulasidhasan
Email: drthulasi86@gmail.com
Vidya, J. Thulasidhasan, and N. V. Prabhu *
Department of Chemistry, School of Arts and Science, Ponnaiyah Ramajayam Institute of Science and Technology (PRIST) Thanjavur, Tamilnadu, India.
THULASIDHASAN J, THEORETICAL INVESTIGATION OF β-CYCLODEXTRIN WITH PYRAZOLINE DERIVATIVE(2021)Journal of Computational Chemistry & Molecular Modeling 5(1)
Semi empirical PM3 calculations were performed upon the inclusion complexation of β-CD with two different pyrazoline derivatives. The negative ∆G and ∆H for the inclusion complexes indicated that the formation of these complexes is exothermic and spontaneous.
H-bonding interactions plays a major role in the CMD1:βCD inclusion process. The theoretical results indicated that the formation of both the inclusion complexes were enthalpy driven process.
Keywords: inclusion complex, Cyclodextrin (CD), molecular modeling.
The synthesis of heterocyclic molecules has wide scope of attentions due to prevalent utility in many biological and medicinal applications. Since their heterocyclic skeleton occur in several natural products, as well as many alkaloids, hormones, vitamins, antibiotics, pharmaceuticals, agro based chemicals, dye stuffs [1], etc., in addition to that the naturally occurring compounds, enormous synthetic heterocycles with significant properties are also recognized [2]. Such type of heterocyclic derivatives afford prospective pharmacophores, which may assemble to supply the high efficient drugs [3]. Similarly these types of available heterocyclic moieties possess good solubility for oral captivation and also bio-compatibility [4]. In accordance to the presence of medicinal properties of the pyrazole moiety, possess some of the therapeutic activities such as anti-inflammatory, antibacterial, antidepressant, anticancer, anti-tuberculosis, antifungal, antioxidant, as well as antiviral agents etc.
Cyclodextrins (CDs) are macrocyclic oligosaccharides made up of six to twelve glucopyranose units linked in a bucket-shaped structure [5]. They shows hydrophobic cavity surrounded by two rims, a wide and a narrow one, composed of primary and secondary hydroxyl groups. By advantage of this arrangement, CDs are able to produce inclusion complexes with a wide variety of hydrophobic organic compounds in aqueous solution. The driving forces leading to complexation are plentiful, varying from hydrophobic to van der Waals and to dipole–dipole interactions [6,7]. Since CDs and their complexes are widely used in synthesis and pharmaceutical sciences, there is currently a great deal of interest in the computational study of their addition complexes.
Because of the experimental limitations, the geometric details and the interactions that stabilize the molecular architecture of pyrazoline derivatives/β-CD inclusion compounds are still poorly understood. For this reason the present study theoretically investigates the interaction between one of the pyrazoline derivative and β-CD molecules by means of PM3 semi-empirical quantum-mechanical calculations, to examine in detail the insertion pathways and to determine the intimate configurations of the inclusion complexes. Bearing in mind that the CD inner cavity cannot fully include the guest molecule, the aim of this work is to identify the part of the molecular cavity that is more suitable for complexation with the pyrazoline derivatives. This study can be useful to predict which of the hydroxyl groups of CDs can be chemically altered in order to avoid the shielding of the cavity during the inclusion process, and, as a consequence, to enhance the systemic pharmacokinetics and bioavailability of the inclusion complexes.
The theoretical calculations were performed with Gaussian 03W. The initial geometry of the selected drug molecules (Fig 1) and β-CD were constructed with Sparton 08 and then optimized by PM3. β-CD was fully optimized by PM3 without any symmetry constraint.
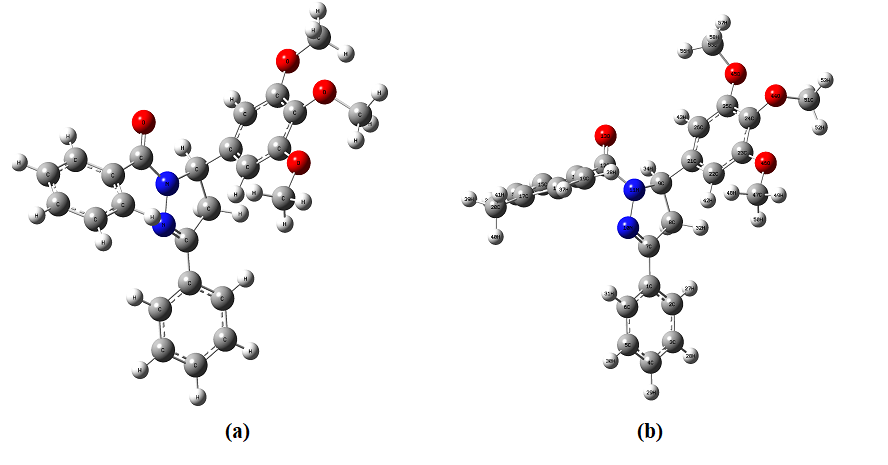
Fig. 3.1. The optimized structure of four different selected drug molecules.
a) 1,3-diphenyl-5-(3,4,5-trimethoxyphenyl)-4,5-dihydro-1H-pyrazole (CMD1)
b) 3-phenyl-1-(p-tolyl)-5-(3,4,5-trimethoxyphenyl)-4,5-dihydro-1H-pyrazole (CMD2)
The inclusion process was simulated by putting the guest in one end of β-CD and then letting it undergo to its cavity. Since the semi-empirical PM3 method proved that to be a strong device within the conformational study of cyclodextrin complexes and has high computational efficiency in calculating the CDs systems [8–15], it was selected to study the inclusion process of β-CD with selected drug molecule in this paper.
HOMO as ionization energy (IE) and LUMO as electron affinity (EA) were used for calculating the electronic chemical potential (μ) which is half of the energy of the HOMO and LUMO:
μ = (EHOMO + ELUMO) /2 (2.1)
The hardness (η) as half of the gap energy between HOMO and LUMO was calculated using the following expression:
Gap = EHOMO – ELUMO (2.2)
η = ELUMO – EHOMO /2 (2.3)
The electrophilicity (ω) of the components were calculated in semiempirical method using the following equation:
ω = μ2/2η (2.4)
The energy, chemical potential, stability, dipole moment, hardness, electrophilicity, HOMO, LUMO, thermodynamic parameters (enthalpy, entropy, free energy), zero point vibrational energy and Mulliken charge of the guest [two different pyrazoline derivatives (Fig. 1)], host (β-CD) and inclusion complexes are summarized. From Fig .2, it shows that the guest molecules formed stable inclusion complexes with β-CD. Interestingly, it observed that the structure of the above drugs: β-CD inclusion complexes were similar to each other.
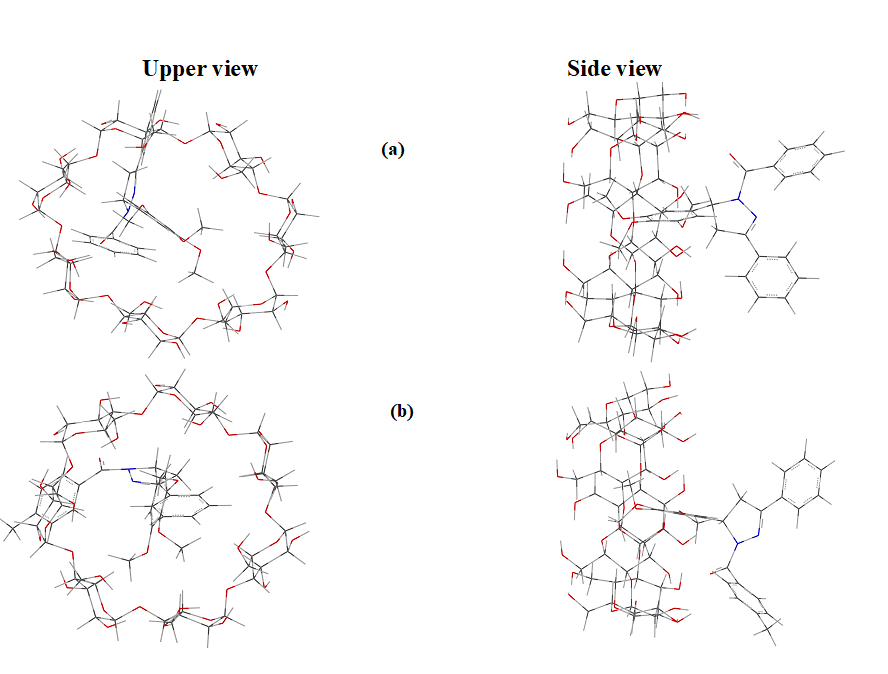
Fig. 2. Axial and equatorial views of low energy structures calculated for the complexes between β-CD and drug molecules.
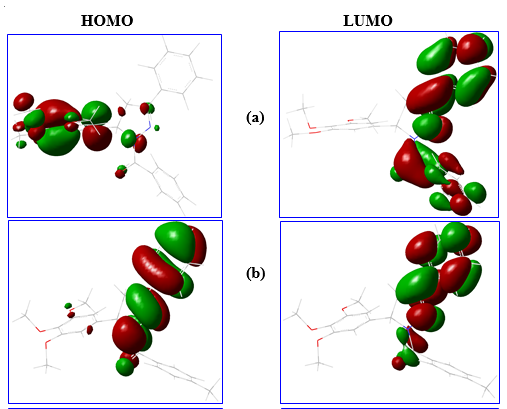
Fig. 3. The HOMO, LUMO energy structure of different drug molecules.
The free energy, enthalpy and entropy for the inclusion complexes were more negative than that of isolated drug and β-CD. The negative ΔG of the inclusion complexes suggest that the inclusion proceeded spontaneously at room temperature. The high negative ΔG value for CMD1: β-CD inclusion complex indicated that this inclusion process is more spontaneous than other complexes.
From the above study we find out that
(i) the stability of the inclusion complexes were same, (ii) both drug molecules were partially encapsulated in to the β-CD cavity, (iii) the negative ∆G and ∆H for the inclusion complexes indicated that the formation of these complexes are spontaneous and weak exothermic, (iv) hydrogen bonding interactions played a major role in the CMD1:β-CD inclusion process while hydrophobic interactions were present in other inclusion complex.
Y. Ju and R. S. Varma, Aqueous N, J. Org. Chem., 71, 135-141, 2006. PMid:16388628
View Article PubMed/NCBID. Za'rate-Za'rate, R. Aguilar, R. I. Herna'ndez-Benitez, E. M. Labarrios, F. Delgado and J. Tamariz, Tetrahedron, 71, 6961-6978, 2015.
View ArticleE. M. Gordon, R. W. Barrett, W. J. Dower, S. P. A. Fodor and M. A. Gallop, J. Med. Chem., 37, 1385-1401, 1994. PMid:8182695
View Article PubMed/NCBIP. D. Leeson and B. Springthorpe, The influence of drug-like concepts on decision making in medicinal chemistry. Nat. Rev. Drug Discovery, 6, 881-890, 2007. PMid:17971784
View Article PubMed/NCBIG, Tiwari.; R. Tiwari, A. K. Rai, J. Pharm. BioAllied Sci. 2010, 2, 72-79. doi:10.4103/0975-7406.67003 PMid:21814436
View Article PubMed/NCBIC. Morari, D. Bogdan, Rom. J. Phys. 2005, 50, 995-1002.
Martin Del Valle, E. M. Process Biochem. 2004, 39, 1033-1046. doi:10.1016/S0032-9592(03) 258-9 00258-9
View ArticleM Shanmugam, J Thulasidhasan, G Venkatesh, V Chidambaranathan, Physics and Chemistry of Liquids 52 (5), 583-600, 2014
View ArticleM Jude Jenita, J Thulasidhasan, N Rajendiran, Journal of Inclusion Phenomena and Macrocyclic Chemistry 79 (3), 365-381, 2014
View ArticleN Rajendiran, T Mohandoss, J Thulasidasan Journal of fluorescence 24 (4), 1003-1014, 2014 PMid:24876092
View Article PubMed/NCBIG Venkatesh, J Thulasidhasan, N Rajendiran Journal of Inclusion Phenomena and Macrocyclic Chemistry 78 (1-4), 225- 237, 2014
View ArticleN Rajendiran, J Thulasidhasan, J Saravanan Spectrochimica Acta Part A 122, 411-421, 2014 PMid:24317267
View Article PubMed/NCBIS Siva, J Thulasidhasan, N Rajendiran Spectrochimica Acta Part A: 115, 559-567, 2013 PMid:23872014
View Article PubMed/NCBIP Thendral, J Thulasidhasan, International Journal of Chemical and Pharmaceutical Sciences 9 (1), 25-33, 2018
N Rajendiran, T Mohandoss, J Thulasidhasan, Physics and Chemistry of Liquids 55 (6), 817-829, 2017