Xiang-Qian Liu
E-mail: lxq0001cn@163.com
© 2019 Sift Desk Journals. All Rights Reserved
VOLUME: 3 ISSUE: 3
Page No: 340-349
Xiang-Qian Liu
E-mail: lxq0001cn@163.com
Jiao Luo1#, Xiang Wang1# , Qin-Peng Zou2, Man-Xia Lu1, Ok-Kyoung Kwon3, Hyeong-Kyu Lee3, Chang-Soo Yook4, Xiang-Qian Liu1*
1School of Pharmacy, Hunan University of Chinese Medicine, Changsha, China, 410208;
2Changsha Broad-Ocean Bio-science and Technique Co.,Ltd., Changsha, China, 410205;
3Korea Research Institute of Bioscience and Biotechnology, Daejeon 341–41,Korea;
4School of Pharmacy, KyungHee University, Seoul 130–701, Korea
# These authors contribute equally to this work and should be considered as the first author
()
Naisheng Bai(nsbai@nwu.edu.cn)
Zhong-Tao Ding(ztding@ynu.edu.cn)
Wioleta Pietrzak(wioleta.pietrzak@umlub.pl)
Xiang-Qian Liu, Separation of Six Lupane-Type Triterpenoid Saponins from Leaves of Acanthopanax gracilistylus by HSCCC with Preparative-HPLC (2018)SDRP Journal of Food Science & Technology 3(3)
High efficiency and less solvent consumption are the essential requirements of high-speed counter current chromatography (HSCCC), especially for the preparation and purification of natural products. In this manuscript, an efficient HSCCC strategy with preparative high performance liquid chromatography (preparative-HPLC) was successfully developed to rapidly separate and purify six lupane-type triterpenoids (including acankoreoside C (1), acangraciliside S (2), acankoreoside B (3), acankoreoside D (4), acantrifoside A (5) and acankoreoside A (6)) from leaves of the Acanthopanax gracilistylus. The effective separation was achieved using ethyl acetate–n-butanol–methanol–water (3:0.3:0.8:4, v/v/v/v) as the two-phase solvent system, in which the mobile phase was eluted at an optimized flow rate of 2.0 mL/min and a revolution speed of 900 rpm. HSCCC preparation was performed on 400 mg of crude sample yielding 5.3 mg of compound 3, 6.4 mg of compound 4, 10.6 mg of compound 5, 35.8 mg of compound 6 with purities of 95.6%, 96.3%, 96.1%, 97.2%, respectively, 17.2 mg of a mixture of compounds 1 and 2, which was further separated by preparative-HPLC yielding 5.9 mg of compound 1, and 4.5 mg of compound 2 with purities of 96.8% and 94.6%, respectively, as determined by HPLC at 210 nm. Their chemical structures were identified by nuclear magnetic resonance (NMR) technology. All compounds were evaluated for their anti-inflammatory activity with lipopolysaccharide (LPS)-induced RAW264.7 cell. The compounds 3 and 4 showed weakly inhibitory effect of nitric oxide (NO) production with low cytotoxicity.
Keywords Acanthopanax gracilistylus, High-speed counter current chromatography (HSCCC), Preparative-HPLC, Nitric oxide (NO), Cytotoxicity
Acanthopanax gracilistylus W.W. Smith (AGS) is an important original plant of Chinese traditional herbs, which is widely distributed in China, such as Hunan, Hubei, Anhui provinces. The leaves of AGS can be used as vegetables to eat for treating skin diseases[1]. Its dried roots and stem barks are listed officially in the Chinese Pharmacopoeia (2015 edition) as Acanthopanax Cortex (named as Wujiapi)[2], which has been used as medicine for the treatment of paralysis, arthritis, rheumatism, lameness, and liver disease[3]. Previous phytochemical investigations of this plant revealed that various natural products from AGS are lignans, triterpenoids, diterpenoids, monoterpenoids, steroids, cerebrosides, and volatile components, which showed diverse biological activities, such as anti-tumor , anti-inflammatory, and liver protective effects[4].
In most of the published reports, the preparation process regarding the isolation of AGS always use silica gel, Sephadex LH-20 or reverse-phase C18 column [5] chromatography repeatedly to obtain pure compounds. But these traditional separation methods are time-consuming and tedious, which makes some sample easily loss. Recently, several literature studies reported the preparative separation of natural products by HSCCC [6–8]. HSCCC being a support-free liquid-liquid partition method, eliminates irreversible adsorption of sample on to the solid support. In many cases, we can acquire compounds with high purity through one-step separation [9], while in other studies enrichment of sample were achieved[10]. The advantages of HSCCC over the conventional separation technique are time-saving, simple process and high recovery [11–15].
In the manuscript, six lupane-type triterpenoid saponins were successfully purified from leaves of the AGS by HSCCC and preparative-HPLC technology, the structure of the six lupane-type triterpenoid saponins, acankoreoside C (1), acangraciliside S (2), acankoreoside B (3), acankoreoside D (4), acantrifoside A (5) and acankoreoside A (6), are shown in the Figure 1. And their anti-inflammatory activities were investigated with LPS-induced RAW264.7 cell.
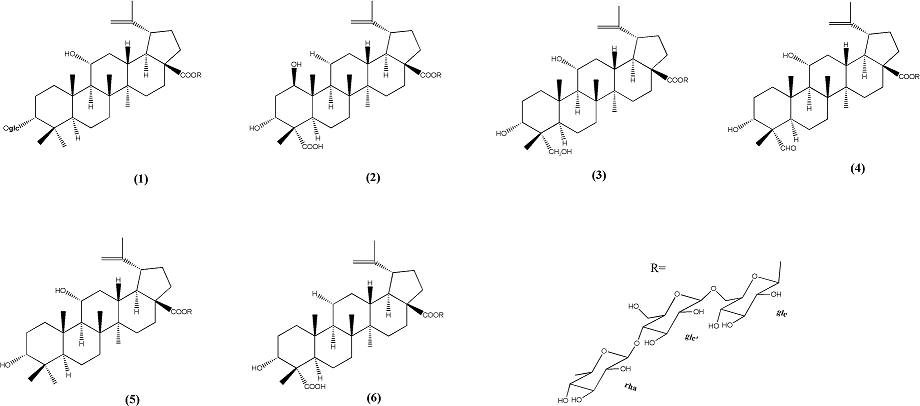
Figure 1: The chemical structures of acankoreoside C (1), acankoreoside S (2), acankoreoside B (3), acankoreoside D (4), acantrifoside A (5) and acankoreoside A (6)
Instrumentation
HSCCC instrument was performed in the present study using a model TBE-300A HSCCC (Shanghai Tauto Biotech Co., Ltd., Shanghai, China). The apparatus was equipped with three multilayer preparative coils connected in series (the diameter of PTFE tube = 1.6 mm, total volume = 300 mL, including the 280 mL separation volume and a 20 mL sample loop). The revolution speed of this instrument was adjustable, ranging from 0 and 1,000 rpm. The two-phase solvent system was pumped into the column by an AKTA prime system (Amersham, USA), and the continuous effluent was monitored with a UV absorbance detector and a DC0506 low constant temperature bath (Shanghai LNB Instrument Co., Ltd). The data were collected with a N2000 chromatography workstation (Zhejiang University Star Information Technology Co., Ltd.; Hangzhou, Zhejiang, China). Preparative-HPLC was performed on an LC-20A liquid chromatograph (Shimadzu Technologies, Kyoto, Japan). Water used was purified by VE-2041-A Ultra-pure water system (Shenzhen Hongsen Environmental Protection Technology Co., Ltd, Shenzhen, China).
Reagents and materials
Methanol and acetonitrile used for HPLC analysis and preparative-HPLC were of chromatographic grade and purchased from Merck, Darmstadt, Germany. All the other organic solvents used for HSCCC were of analytical grade and purchased from Tianjin Hengxing Chemical Preparation Co., Ltd (Tianjin, China). Water used was purified by Ultra-pure water system.
The leaves of AGS were collected in Yuanling, Hunan province of China, in July 2015, and were botanically identified by Professor Xiang-Qian Liu, the corresponding author of this manuscript. A voucher specimen has been deposited in the Herbarium of Hunan University of Chinese Medicine, Hunan, China (No. 20150718).
Preparation of the crude sample
The dried leaves of AGS (600 g) were ground into powder and extracted three times by hot methanol (3 × 3 L) through heating reflux at 65 °C. The combined methanol extract was evaporated under reduced pressure to obtain a residue (120 g), which was dissolved in water and partitioned with petroleum ether (PE, 60-90 °C), ethyl acetate, and n-butanol, successively giving PE layer (3.18 g), ethyl acetate layer (28.23 g), and n-butanol layer (25.04 g) after removal of the solvent in vacuo. The n-butanol layer was stored in a refrigerator (4 °C) for the subsequent HSCCC and preparative-HPLC separation.
Selection of the two-phase solvent system
The two-phase solvent system was selected according to the partition coefficients (K) of the target components. The K values were determined as follows: a suitable amount of crude sample was added into pre-equilibrated two-phase solvent system that volume of each phase was more than 5 mL and then mixed thoroughly[16]. The same volume of the upper and the lower phase was evaporated to dryness. The residues were diluted in 2.5 mL of methanol and then analyzed by HPLC. The partition coefficient (K) were calculated by the ratio of the peak area obtained from the upper phase to that obtained from lower phase.
Preparation of two-phase solvent system and sample solutions
For HSCCC separation, a two-phase solvent system composed of ethyl acetate–n-butanol–methanol–water (3:0.3:0.8:4, v/v/v/v) was used. The two-phase solvent system was thoroughly equilibrated in a separation funnel at room temperature. The upper phase and the lower phase were separately degassed by sonication for 25 min prior to HSCCC separation. The HSCCC sample was prepared by dissolving 400 mg of the dried crude sample in 20 mL of the lower phase.
HSCCC and preparative-HPLC separation procedure
HSCCC separation was performed as follows: the multilayer column was first completely filled with the upper (stationary) phase[17]. Then, the apparatus was rotated at 900 rpm in the forward direction. The lower phase was pumped into the head of the column at the flow rate of 2.0 mL/min when the revolution velocity was smooth. After reaching hydrodynamic equilibrium, as indicated by the emergence of the mobile phase front, 20 mL of the sample solution was injected into the column through the injection valve. The effluent from the tail end of the column was continuously monitored by a UV detector at 210 nm, and the chromatogram was recorded. Each peak fraction was collected according to the chromatogram and analyzed by HPLC. All fractions of the same peak were combined and evaporated to dryness under reduced pressure.
The preparative-HPLC separation was performed as follows: CST C18 column (300 mm×30 mm, 10 μm); the solvent system consisted of acetonitrile–water (23:77, v/v); the eluent was pumped at 20 mL/min (monitored at 210 nm) and all injection volume was 2 mL. The peak fractions were collected according to the elution profile.
HPLC analysis and identification of separated compounds
In this manuscript, the HPLC was used for analyzing the composition of the dried crude samples and determining the purities of the separated compounds. HPLC analyses of the crude sample and each peak fraction obtained from HSCCC were performed with a Promosil C18 column (5 μm, 4.6×250 mm) at a column temperature of 30 °C. The binary mobile phase consisted of acetonitrile and water in a gradient as follows: 0–15 min, 22–30% acetonitrile; 15–21 min, 30% acetonitrile; 21–35 min, 30–40% acetonitrile; 35–45 min, 40–22% acetonitrile; 45–50 min, 22% acetonitrile. All solvents were filtered through 0.45 μm filter prior to use. The flow rate was set to 1.0 mL/min, and the detector wavelength was 210 nm. Identification of separated compounds was carried out by NMR.
MTT assay for cell viability
The viability of RAW 264.7 cells was determined by analyzing the reduction of 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) to formazan. Cells were cultured in the 96 well plates at a density 1×104 per well for 4 h. After incubated at 37 °C in a humidified atmosphere containing 5% CO2 for 24 h, Cell viability was determined by adding 10 μL of MTT (5 mg/mL) to the medium and incubated for 4 h. Finally, the supernatant was removed and the formazan crystals were dissolved in 100 μL of dimethyl sulfoxide (DMSO) and reacted for 10 min. Absorbance was measured at 570 nm.
Nitric oxide (NO) assay
RAW 264.7 cells (5×104 cells per well in 96 well plates) were treated with 10 μL test samples for 1 h, and incubated at 37 °C in a humidified atmosphere containing 5% CO2 before exposure to 0.5 μg/mL of LPS. After 24 h incubation, NO productions indirectly determined by measuring the stable NO catabolite nitrite in medium by Griess reaction. In brief, an aliquot of each supernatant (100 μL) was mixed with the same volume of Griess reagent for 10 min at RT. The absorbance was measured at 540 nm using an enzyme-linked immunosorbent assay (ELISA) plate reader.
HPLC analysis of the crude sample
As shown in Figure 2, the HPLC chromatogram of the crude sample from the leaves of AGS showed compounds 1–6.
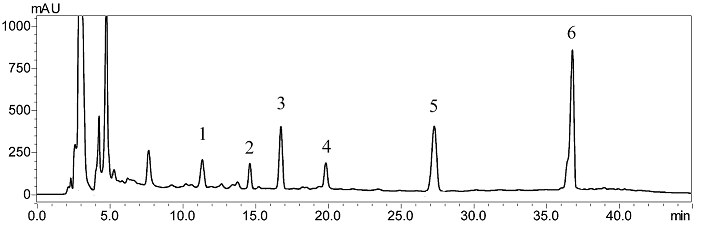
Figure 2: HPLC chromatogram of the crude sample from the leaves of AGS. HPLC Conditions: Promosil C18 column (5 μm, 4.6×250 mm); mobile phase, acetonitrile and water in gradient mode (acetonitrile: 0–15 min, 22–30%; 15–21 min, 30%; 21–35 min, 30–40%; 35–45 min, 40–22%; 45–50 min, 22%); flow rate, 1.0 mL/min; column temperature, 30 °C; detection wavelength, 210 nm.
Selection of HSCCC solvent system
Successful separation by HSCCC requires the careful selection of a suitable two-phase solvent system. The two-phase solvent system was selected according to the partition coefficients (K-values) of the target compound. K-values were obtained and analyzed by the liquid–liquid extraction experiments and HPLC analysis. K-values of the target compounds should be in the range of 0.5–2.0 and the separation factor between two compounds ought to be >1.5[18–21].
According to the polarity of the target compounds, ethyl acetate–n-butanol–methanol–water at different volume ratios of 3:0.8:1:4, 3:0.5:1:4, 3:0.3:1:4, 3:0.3:0.8:4 and 3:0.3:0.5:4, respectively, were tested. As shown in Table 1. when ratios of 3:0.8:1:4 and 3:0.5:1:4 were used as the two-phase solvent systems, their K-values were too large, with which target compounds would be eluted in an excessively broad peak with long elution time. When ratio of 3:0.3:0.5:4 was used, the K-values were too small and the target compounds could not be separated. The composition of 3:0.3:1:4 and 3:0.3:0.8:4 both provided the suitable K-values and the separation factor. However, the ratio of 3:0.3:1:4 provided K6=1.59 for the compound 6, compared to the ratio of 3:0.3:0.8:4 provided K6=1.08. Therefore, the ratio of 3:0.3:0.8:4 had a shorter time and relatively good peak. Therefore, ethyl acetate–n-butanol–methanol–water at a volume ratio of 3:0.3:0.8:4(v/v/v/v) was selected for the HSCCC separation process.
Table 1. The partition coefficient (K) values of the compounds 3–6 at different ratio of volume in ethyl acetate–n-butanol–methanol–water solvent system
|
ethyl acetate– n-butanol–methanol–water (V/V/V/V) |
the partition coefficient (K) |
|||
|
compound 3 |
compound 4 |
compound 5 |
compound 6 |
|
|
3:0.8:1:4 |
1.03 |
1.58 |
2.65 |
3.09 |
|
3:0.5:1:4 |
0.61 |
1.08 |
1.81 |
1.89 |
|
3:0.3:1:4 |
0.44 |
0.61 |
1.41 |
1.59 |
|
3:0.3:0.8:4 |
0.26 |
0.54 |
0.91 |
1.08 |
|
3:0.3:0.5:4 |
0.06 |
0.25 |
0.57 |
0.72 |
HSCCC separation
400 mg of the crude sample extract was dissolved in 20 mL of lower phase and separated according to the procedure described above. The HSCCC fractions were analyzed by HPLC and their absorbance were measured at 210 nm to draw elution curves. Based on HPLC analysis and the elution curve of the preparative HSCCC (Figure 3), collected fractions were combined into different pooled fractions. Compounds 3–6 were obtained and their HPLC chromatograms were shown in Figure 5 (C–F). As determined by HPLC, their purities were 95.6, 96.3, 96.1, 97.2%, respectively. The retention of the stationary phase was 46.7%.
Preparative-HPLC separation
After HSCCC separation, Fr.1 and Fr.2–5 were concentrated, yielding a mixture (containing compounds 1 and 2) and compounds 3–6, respectively. Then, the mixture (containing compounds 1 and 2) was separated by preparative-HPLC (Figure 4), yielding 5.9 mg of compound 1, and 4.5 mg of compound 2. Based on the HPLC analysis, the purities were 96.8 and 94.6%, respectively. As shown in Figure 5 (A–B).
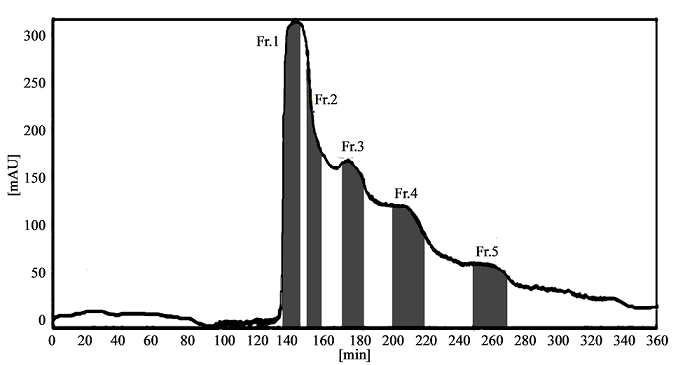
Figure 3: Preparative HSCCC separation of the crude sample from AGS. Stationary phase: upper organic phase; mobile phase: lower aqueous phase; flow rate: 2.0 mL/min; revolution speed: 900 rpm; column temperature: 25 °C; crude sample: 400 mg dissolved in 20 mL mixture solution of lower phase of the solvent system; solvent system: ethyl acetate–n-butanol–methanol–water (3:0.3:0.8:4, v/v/v/v); retention of the stationary phase: 46.7%; detector:210 nm. Fr.1 = compound 1+compound 2; Fr.2 = compound 3; Fr.3 = compound 4; Fr.4 = compound 5; Fr.5 = compound 6.
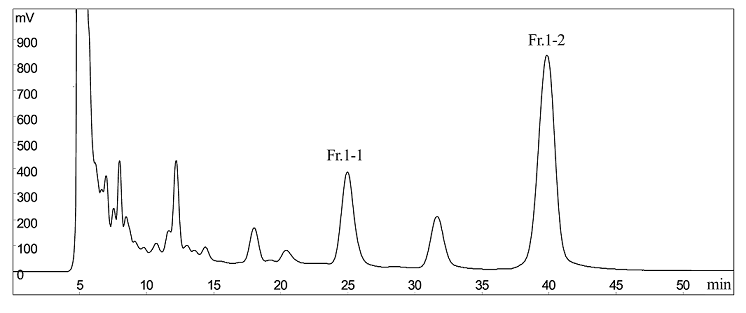
Figure 4: preparative-HPLC chromatogram of Fr.1 in HSCCC. preparative-HPLC condition: CST C18 column (300 mm×30 mm , 10 μm); mobile phase: acetonitrile : water (23:77, v/v); flow rate:20 mL/min; (monitored at 210 nm) .Fr.1–1 = compound 1, Fr.1–2 = compound 2.
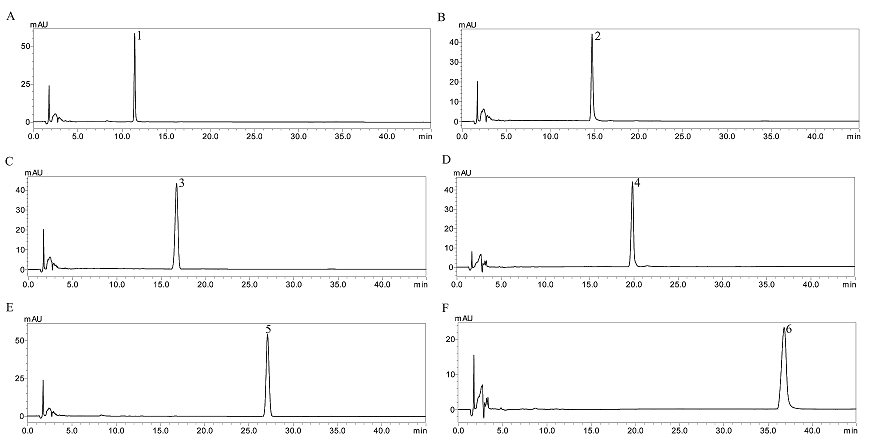
Figure 5: HPLC chromatograms. A = compound 1; B = compound 2; C = compound 3; D = compound 4; E = compound 5; F = compound 6. HPLC conditions: Promosil C18 column (5 μm, 4.6×250 mm); mobile phase, acetonitrile and water in gradient mode (acetonitrile: 0–15 min, 22–30%; 15–21 min, 30%; 21–35 min, 30–40%; 35–45 min, 40–22%; 45–50 min, 22%); flow rate, 1.0 mL/min; column temperature, 30 °C; detection wavelength, 210 nm.
Structural identification
The chemical structure of six compounds separated by HSCCC and preparative-HPLC were identified according to their NMR data.
Acankoreoside C (1):white powder; 13C-NMR (125MHz, CD3OD-d4): 36.62 (C-1), 22.12 (C-2), 82.33 (C-3), 38.35 (C-4), 50.91 (C-5), 19.09 (C-6), 36.21 (C-7), 43.46 (C-8), 56.19 (C-9), 40.24 (C-10), 70.69 (C-11), 38.38 (C-12), 37.59 (C-13), 43.89 (C-14), 30.69 (C-15), 32.86 (C-16), 57.89 (C-17), 50.22 (C-18), 48.17 (C-19), 151.32 (C-20), 31.60 (C-21), 37.47 (C-22), 29.65 (C-23), 23.14 (C-24), 17.18 (C-25), 17.84 (C-26), 15.06 (C-27), 175.1 (C-28), 110.75 (C-29), 20.19 (C-30), 95.30 (C-1glc), 73.73 (C-2glc), 78.38 (C-3glc), 71.03 (C-4glc), 77.72 (C-5glc), 69.54 (C-6glc), 104.4 (C-1glc'), 75.21 (C-2glc'), 76.68 (C-3glc'), 78.23 (C-4glc'), 76.84 (C-5glc'), 61.80 (C-6glc'), 102.93 (C-1rha), 72.15 (C-2rha), 72.40 (C-3rha), 74.01 (C-4rha), 70.69 (C-5rha), 17.84 (C-6rha), 101.59 (C-1glc''), 75.27 (C-2glc''), 79.67 (C-3glc''), 72.05 (C-4glc''), 78.06 (C-5glc''), 63.02 (C-6glc''). The 13C-NMR data for compound 1 agree with the literature data[22] corresponding to acankoreoside C.
Acangraciliside S (2): white powder; 13C-NMR (125MHz, CD3OD-d4): 76.33 (C-1), 36.79 (C-2), 74.14 (C-3), 52.24 (C-4), 46.11 (C-5), 22.17 (C-6), 35.09 (C-7), 42.96 (C-8), 53.06 (C-9), 44.47 (C-10), 24.82 (C-11), 26.94 (C-12), 39.13 (C-13), 43.82 (C-14), 30.86 (C-15), 32.95 (C-16), 57.93 (C-17), 50.6 (C-18), 48.36 (C-19), 151.77 (C-20), 31.55 (C-21), 37.68 (C-22), 182.6 (C-23), 18.08 (C-24), 13.17 (C-25), 17.14 (C-26), 15.1 (C-27), 176.4 (C-28), 110.41 (C-29), 19.49 (C-30), 95.26 (C-1glc), 74 (C-2glc), 79.51 (C-3glc), 70.95 (C-4glc), 78.06 (C-5glc), 69.55 (C-6glc), 104.56 (C-1glc'), 75.32 (C-2glc'), 76.71 (C-3glc'), 78.28 (C-4glc'), 76.89 (C-5glc'), 61.90 (C-6glc'), 102.92 (C-1rha), 72.16 (C-2rha), 72.44 (C-3rha), 73.75 (C-4rha), 70.64 (C-5rha), 17.84 (C-6rha). The 13C-NMR data for compound 2 agree with the literature data[23] corresponding to acangraciliside S.
Acankoreoside B (3):white powder; 13C-NMR data (125 MHz, pyrindine-d5): 36.30 (C-1), 27.51 (C-2), 76.32 (C-3), 41.52 (C-4), 44.22 (C-5), 18.66 (C-6), 35.77 (C-7), 43.15 (C-8), 56.60 (C-9), 40.01(C-10), 70.18 (C-11), 38.66 (C-12), 37.78 (C-13), 43.29 (C-14), 30.38 (C-15), 32.64 (C-16), 57.29 (C-17), 49.83 (C-18), 47.54 (C-19), 150.79 (C-20), 31.25 (C-21), 37.10 (C-22), 72.28 (C-23), 18.74 (C-24), 17.48 (C-25), 18.11 (C-26), 15.15 (C-27), 175.35 (C-28), 110.57 (C-29), 19.85 (C-30), 95.66 (C-1glc), 74.35 (C-2glc), 79.06 (C-3glc), 71.21 (C-4glc), 78.38 (C-5glc), 69.79 (C-6glc), 105.48 (C-1glc'), 75.67 (C-2glc'), 76.81 (C-3glc'), 78.57 (C-4glc'), 77.53 (C-5glc'), 61.66 (C-6glc'), 103.08 (C-1rha), 72.94 (C-2rha), 73.13 (C-3rha), 74.40 (C-4rha), 70.68 (C-5rha), 18.89 (C-6rha). The 13C-NMR data for compound 3 agree with the literature data[24] corresponding to acankoreoside B.
Acankoreoside D (4): white powder; 13C-NMR data (125MHz, pyrindine-d5): 35.56 (C-1), 27.51 (C-2), 73.44 (C-3), 53.35 (C-4), 44.60 (C-5), 21.73 (C-6), 35.77 (C-7), 43.10 (C-8), 56.30 (C-9), 39.39 (C-10), 70.06 (C-11), 38.66 (C-12), 37.78 (C-13), 43.73 (C-14), 30.38 (C-15), 32.64 (C-16), 57.27 (C-17), 49.83 (C-18), 47.54 (C-19), 150.78 (C-20), 31.25 (C-21), 37.10 (C-22), 210.50 (C-23), 18.74 (C-24), 17.19 (C-25), 18.16 (C-26), 15.33 (C-27), 175.35 (C-28), 110.57 (C-29), 19.85 (C-30), 95.66 (C-1glc), 74.35 (C-2glc), 78.57 (C-3glc), 71.21 (C-4glc), 77.53 (C-5glc), 69.79 (C-6glc), 105.48 (C-1glc'), 75.67 (C-2glc'), 76.81 (C-3glc'), 79.06 (C-4glc'), 78.38 (C-5glc'), 61.66 (C-6glc'), 103.08 (C-1rha), 72.94 (C-2rha), 73.13 (C-3rha), 74.40 (C-4rha), 70.68 (C-5rha), 18.89 (C-6rha). The 13C-NMR data for compound 4 agree with the literature data[24] corresponding to acankoreoside D.
Acantrifoside A (5): white powder; 13C-NMR data (125 MHz, pyrindine-d5): 36.54 (C-1), 27.24 (C-2), 75.64 (C-3), 38.96 (C-4), 49.94 (C-5), 18.94 (C-6), 36.24 (C-7), 43.02 (C-8), 56.54 (C-9), 40.24 (C-10), 70.25 (C-11), 38.64 (C-12), 37.81 (C-13), 43.32 (C-14), 30.44 (C-15), 32.65 (C-16), 57.37 (C-17), 49.86 (C-18), 47.45 (C-19), 150.86 (C-20), 31.27 (C-21), 37.21 (C-22), 30.28 (C-23), 23.34 (C-24), 17.26 (C-25), 18.08 (C-26), 15.14 (C-27), 175.46 (C-28), 110.53 (C-29), 19.82 (C-30), 95.68 (C-1glc), 74.37 (C-2glc), 78.66 (C-3glc), 71.25 (C-4glc), 77.47 (C-5glc), 69.77 (C-6glc), 105.49 (C-1glc'), 75.68 (C-2glc'), 76.86 (C-3glc'), 79.04 (C-4glc'), 78.45 (C-5glc'), 61.75 (C-6glc'), 103.14 (C-1rha), 72.81 (C-2rha), 73.06 (C-3rha), 74.45 (C-4rha), 70.68 (C-5rha), 18.87 (C-6rha). The 13C-NMR data for compound 5 agree with the literature data[22] corresponding to acantrifoside A.
Acankoreoside A (6): white powder; 13C-NMR (125MHz, pyrindine-d5): 33.64 (C-1), 26.46 (C-2), 73.14 (C-3), 51.95 (C-4), 45.74 (C-5), 21.96 (C-6), 34.85 (C-7), 42.06 (C-8), 51.28 (C-9), 37.67 (C-10), 21.24 (C-11), 26.36 (C-12), 38.84 (C-13), 43.02 (C-14), 31.28 (C-15), 32.78 (C-16), 57.37 (C-17), 50.09 (C-18), 47.78 (C-19), 150.94 (C-20), 31.43 (C-21), 37.03 (C-22), 179.7 (C-23), 18.61 (C-24), 17.36 (C-25), 16.95 (C-26), 15.29 (C-27), 175.3 (C-28), 110.77 (C-29), 19.84 (C-30), 95.65 (C-1glc), 74.46 (C-2glc), 78.62 (C-3glc), 71.34 (C-4glc), 78.49 (C-5glc), 69.85 (C-6glc), 105.48 (C-1glc'), 75.77 (C-2glc'), 76.85 (C-3glc'), 79.07 (C-4glc'), 78.48 (C-5glc'), 61.77 (C-6glc'), 102.94 (C-1rha), 72.85 (C-2rha), 73.06 (C-3rha), 74.48 (C-4rha), 70.67 (C-5rha), 18.82 (C-6rha). The 13C-NMR data for compound 6 agree with the literature data[24] corresponding to acankoreoside A.
Results of cytotoxicity and inhibition NO
The cytotoxicity and inhibition of production of NO of compounds 1–6 from AGS were investigated on LPS-induced RAW264.7 cell. As shown in Table 2, the production of NO was down-regulated weakly when the concentration of compounds 3 and 4 was 20 μg/mL with the inhibition as 4.18% and 14.32%, respectively. Compounds 1, 2, 5, 6 had no inhibition of production of NO. Compound 1 showed moderate cytotoxicity when the concentration of compound 1 was 20 μg/mL with the cell viability as 55.61% and compounds 2, 3, 4, 5, 6 had no influence on cell viability.
Table 2. Inhibitory effects of compounds 1-6 against LPS-induced NO production and cell viability in RAW 264.7cell
|
compounds |
inhibition |
cell viability |
|
acankoreoside C(1) |
- |
55.61% |
|
acankoreoside S(2) |
- |
101.32% |
|
acankoreoside B(3) |
4.18% in 20 μg/ml |
106.86% |
|
acankoreoside D(4) |
14.32% in 20 μg/ml |
96.64% |
|
acantrifoside A(5) |
- |
103.67% |
|
acankoreoside A(6) |
- |
92.6% |
In summary, we have known that the combination of application of HSCCC and preparative-HPLC is a very powerful technique for natural products. In present work, an efficient separation of six lupane-type triterpenoid saponins from the leaves of AGS was achieved by HSCCC and preparative-HPLC for the first time. Compared to reported in the literature on separation of lupane-type triterpenoid saponins from the leaves of AGS, the method of HSCCC and preparative-HPLC was fewer time-consuming, one-step separation, higher recovery and higher purity. Six lupane-type triterpenoid saponins: acankoreoside C (1), acangraciliside S (2), acankoreoside B (3), acankoreoside D (4), acantrifoside A (5) and acankoreoside A (6) were yielded in HSCCC and preparative-HPLC with purities of 95.6%, 96.3%, 96.1%, 97.2%, 96.8%, and 94.6%, respectively. All these triterpenoid saponins were tested cell viability and anti-inflammatory activity with LPS-induced RAW264.7 cell. Among those compounds, compounds 3 and 4 showed weakly inhibitory effect of NO production with lower cytotoxicity, and compound 1 only showed moderate cytotoxicity activity against RAW264.7 cell. In the literature AGS has diverse biological activities, and is rich in lupane-type triterpenoid saponins, thus it is necessary to be investigated other bioactivity of these compounds in the future. Therefore, this manuscript provided an efficient, convenient and economical method for the separation and purification of effective compounds from natural products.
The present study was supported by a grant from the Natural Science Foundation of Hunan Province, China (grant no. 11JJ2042) and the Key Projects of Changsha City Science and Technology Bureau (kq1701119), Hunan Provincial Innovation Foundation for Postgraduate ( CX2017B444).
New medical school of Jiangsu province. Great Dictionary of Chinese Medicine [M]. China: Shanghai scientific & technical publishers, 1977: 380
Zou QP, Liu XQ, Huang JJ, Yook CS, Whang WK, Lee HK, Kwon OK (2017) Inhibitory effects of lupane-type triterpenoid saponins from the leaves of Acanthopanax gracilistylus on lipopolysaccharide-induced TNF-α, IL-1β and high-mobility group box 1 release in macrophages. Mol Med Rep 16(6):9149-9156 PMid:29039503
View Article PubMed/NCBILi XJ, Dai L, Li Z, Zhang XD, Liu XQ, Zou QP (2015) Anti-inflammatory Activities of Lupane-triterpenoids In Vitro and Their Phytochemical Fingerprinting from Leaves of Acanthopanax gracilistylus. Natural product sciences 21(2): 104-110
Dai L, Liu XQ, Xie X, Liu HY (2014) Characterization of stereostructure by X-ray and technology of extracting in combination hydrolysis in situ of acankoreanogenin from leaves of Acanthopanax gracilistylus W. W. Smith. J. Cent. South Univ 21(8):3063-3070
View ArticleLiu XQ, Chang SY, Park SY; Nohara T, Yook CS (2002) A New Lupane-Triterpene Glycoside from the Leaves of Acanthopanax gracilistylus. Arch Pharm Res 25(6): 831-836 PMid:12510834
View Article PubMed/NCBIYang MX, Liang YG, Chen HR, Huang YF, Gong HG, Zhang TY (2018) Isolation of Flavonoids From Wild Aquilaria sinensis Leaves by an Improved Preparative High-Speed Counter-Current Chromatography Apparatus. J Chromatogr Sci 56(1):18-24 PMid:28977348
View Article PubMed/NCBIJiang WH, Shan H, Song JY, Lü HT (2017) Separation and Purification of Ombuoside from Gynostemma Pentaphyllum by Microwave-Assisted Extraction Coupled with High-Speed Counter-current Chromatography. J Chromatogr Sci 55(1):69-74 PMid:27993866
View Article PubMed/NCBIGu B, Zhang Y, Ding L, He S, Wu B, Dong J, Zhu P, Chen J, Zhang J, Yan X (2015) Preparative Separation of Sulfur-Containing Diketopiperazines from Marine Fungus Cladosporium sp. Using High-Speed Counter-Current Chromatography in Stepwise Elution Mode. Mar Drugs 13(1): 354-365 PMid:25584683
View Article PubMed/NCBILiu Y, Wang P, Chen T, Jia J, Sun J, You JM, Li YL (2014) One-Step Isolation and Purification of Four Xanthone Glycosides from Tibetan Medicinal Plant Halenia elliptica by High-Speed Counter-Current Chromatography. Sep Sci Technol 49(7): 1119-1124
View ArticleHe YF, Wang XY, Suo YR, Ding CX, Wang HL (2016) Efficient Protocol for Isolation of Rhaponticin and Rhapontigenin with Consecutive Sample Injection from Fenugreek (Trigonella foenum-graecum L.) by HSCCC. J Chromatogr Sci 54(3): 479-485 PMid:26598549
PubMed/NCBIGeng P, Fang YT, Xie RL, Hu WL, Xi XG, Chu Q, Dong GL, Shaheen N, Wei Y (2017) Separation of phenolic acids from sugarcane rind by online solid-phase extraction with high-speed counter-current chromatography. J Sep Sci 40(4):991-998 PMid:27943588
View Article PubMed/NCBISun YS, Hou ZG, Liu ZB, Wang JH (2016) Ionic Liquid-Based Ultrasonic-Assisted Extraction of Forsythosides from the Leaf of Forsythia suspensa (Thunb.) Vahl and Subsequent Separation and Purification by High-Speed Counter-Current Chromatography. J Chromatogr Sci 54(8): 1445-1452 PMid:27165571
View Article PubMed/NCBIZhou YQ, Wang CM, Wang RB, Lin LG, Yin ZQ, Hu H, Yang Q, Zhang QW (2017) Preparative separation of four sesquiterpenoids from Curcuma longa by high-speed counter-current chromatography. Sep Sci Technol 52(3): 497-503
View ArticleLu QX, Sun YR, Shu YY, Tan SC, Yin L, Guo YR, Tang L (2016) HSCCC Separation of the Two Iridoid Glycosides and Three Phenolic Compounds from Veronica ciliata and Their in Vitro Antioxidant and Anti-Hepatocarcinoma Activities. Molecules 21(9): 1-13 PMid:27649125
View Article PubMed/NCBISun YJ, Pei LX, Wang KB, Sun YS, Wang JM, Zhang YL, Gao ML, Ji BY (2015) Preparative Isolation of Two Prenylated Biflavonoids from the Roots and Rhizomes of Sinopodophyllum emodi by Sephadex LH-20 Column and High-Speed Counter-Current Chromatography. Molecules 21(10): 1-13
View ArticleZhang L, Yue HL, Zhao XH, Li J, Shao Y (2015) Separation of Four Phenylpropanoid Glycosides from a Chinese Herb by HSCCC. J Chromatogr Sci 53: 860–865 PMid:25410625
View Article PubMed/NCBIZhou PJ, Luo QJ, Ding LJ, Fang F, Yuan Y, Chen JJ, Zhang JR, Jin HX, He S (2015) Preparative Isolation and Purification of Lignans from Justicia procumbens Using High-Speed Counter-Current Chromatography in Stepwise Elution Mode. Molecules 20(4): 7048-7058 PMid:25903362
View Article PubMed/NCBILv HH, Zhou WN, Wang XY, Wang ZH, Suo YR, Wang HL (2016) Extraction and Separation of Vitisin D, Ampelopsin B and cis-Vitisin A from Iris lactea Pall. var. chinensis (Fisch.) Koidz by Alkaline Extraction-Acid Precipitation and High-Speed Counter-Current Chromatography. J Chromatogr Sci 54(5): 744-751 PMid:26847919 PMCid:PMC4890446
View Article PubMed/NCBISun Y, Yu Z, Duan W, Fang L, Xu S, Wang X (2011) Isolation and purification of seven lignans from Magnolia sprengeri by high-speed counter-current chromatography. J Chromatogr B 879(31): 3775-3779. PMid:22080044
View Article PubMed/NCBIDuan WJ, Bai AY, Lin XJ, Fang L, Wang X (2014) Isolation and Purification of Highly Polar Antioxidants from Chirita longgangensis by Combination of Macroporous Resin and HSCCC. Chromatographia 77(9-10): 707-713
View ArticleYe XL, Cao D, Song FY, Fan GR, Wu FH (2016) Preparative separation of nine flavonoids from Pericarpium Citri Reticulatae by preparative-HPLC and HSCCC. Sep Sci Technol 51(5): 807-815
View ArticleLiu XQ, Chang SY, Yook CS (2006) Lupane-triterpenoids from the leaves of Acanthopanax gracilistylus. Journal of Lanzhou University (Natural Sciences) 42(4): 86-91
Li XJ, Zou QP, Wang X, Kim KW, Lu MF, Ko SK, Yook CS, Kim YC, Liu XQ (2018) Lupane Triterpenes from the Leaves of Acanthopanax gracilistylus. Molecules. http: //dx. doi. org/ 10.3390/ molecules 23010087
Zou QP, Liu XQ, Lee HK, Oh OJ (2011) Lupane-triterpenoids from the methanol extracts of leaves of Acanthopanax gracilistylus W.W. Smith. Journal of Lanzhou University (Natural Sciences) 47(6): 120-126