Fatemeh Ghorbania
Email: tima.ghorbani@gmail.com
© 2019 Sift Desk Journals. All Rights Reserved
VOLUME: 5 ISSUE: 3
Page No: 182-198
Fatemeh Ghorbania
Email: tima.ghorbani@gmail.com
Fatemeh Ghorbania,b*, Masoomeh Ghorbanic, Arezou Ghahghaeeb
a Medical Biology Research Center, Health Technology Institute, Kermanshah University of Medical Sciences, Kermanshah, Iran
bDepartment of Biology, Faculty of Sciences, University of Sistan and Baluchestan, Zahedan, Iran, cDepartment of Agronomy and plant breeding, Faculty of Sciences, Razi University, Kermanshah, Iran
Fatemeh Ghorbani, The Inhibitory Effects of Nucleosides, Nicotinamide Adenine Dinucleotide, Adenosine 5'-Triphosphate, Inosine, Nicotinamide Riboside and Nicotinamide Mononucleotide Against α-Amylase and α-Glucosidase Enzymes (2020) Journal of Food Science & Technology 5(4) pp:182-198
Diabetes is a group of metabolic disorders characterized by a high blood sugar level over a prolonged period of time. Inhibition of carbohydrate hydrolyzing enzymes leads to decrease in the absorption of glucose which is considered as one of the effective managements of diabetes mellitus. Vegetable, fruit, milk and fish are good sources of nucleosides and inosine (INO), nicotinamide riboside (NR) and nicotinamide mononucleotide (NMN) with versatile health benefits. The well-adapted structural features of these compounds for the inhibition/activation of enzymes include several available hydrogen bond (H-bond) acceptors and donors, flexible backbone and hydrophobic nature. The substrates of α-amylase (α-Amy) and α-Glucosidase (α-Glu), known as key absorbing enzymes, have functional groups (OH groups) resembling nucleosides. Therefore, the present study was conducted to evaluate the inhibitory properties of nucleosides against αAmy and α-Glu. The median inhibition concentration (IC50) values for α-Glu in the presence of adenosine (ADN), adenosine triphosphate (AMP), NR, INO, adenosine triphosphate (ATP), nicotinamide adenine dinucleotide (NAD), Adenosine diphosphate (ADP)-ribose, ADP-glucose and NMN were determined 208.6±3.8, 254.1±5.2, 177.7±4.8, 192.1±5.2, 215.9±2.7, 65.4±1.3, 63.4±2.2, 75.6±4.2 and 196.1±2.6, respectively. The IC50 values α-Amy in the presence of ADN, AMP, NR, INO, ATP, NAD, ADP-ribose, ADP-glucose and NMN were determined 145.3±2.4, 202.3±3.9, 127.7±4.8, 163.5±3.6, 185.3±1.2, 80.4±2.8, 64.8±4.7, 51.1±1.6 and 166.5±1.4, respectively. Moreover, the Ki values of NAD were calculated as 13.8±0.8 and 18.6±2.4 µM for α-Glu and α-Amy in a competitive-mode and noncompetitive -mode inhibition. In addition, to communicate with the active site of α-Glu and α-Amy respectively, NR presented a binding energy of -7.8 and -6.8 kcal/mol, INO -7.3 and -6.9, ATP -8.3 and -7.3, NAD -10.0 and -8.5, ADP-ribose -8.7 and -7.4, ADP-glucose -8.9 and -7.6, cAMP -6.6 and -6.3 and NMN -6.8 and -7.0 kcal/mol. These antioxidant inhibitors may be potential anti-diabetic drugs, not only to reduce glycemic index, but also to limit the activity of the major reactive oxygen species (ROS) producing pathways.
Key words: Nucleosides, NAD, hydrolyzing enzymes, enzyme inhibition, hyperglycemia
Diabetes mellitus (DM) is a chronic disease with the characteristics of abnormal insulin resistance and glucose tolerance [1]. There are some complications with DM including metabolic syndrome, heart disease, renal function recession, and blindness [2]. Hyperglycemia is one of the most important metabolic complications in type-2 DM (T2D) which lead to the development of nephropathy, retinopathy, neuropathy, heart and blood vessel diseases [3]. Preventing of hydrolyzing of carbohydrates is a novel treatment method and alternative medicine for treating T2D through delay in the digestion of carbohydrates ultimately reducing the rate of glucose absorption [4]. α-Amy and α-Glu are hydrolyzing enzymes secreted and placed in small intestine and they are responsible for hydrolyzing dietary carbohydrates.
Acarbose (ACR) is the most common drug which used as an α-Amy/α-Glu inhibitor for T2D. However, it has been indicated that this drug has numerous adverse impacts including abdominal distention, diarrhea, flatulence, and pneumatosis cystoides intestinalis [5]. Fermentation of undigested carbohydrates by colonic bacteria is considered as one of the most important reasons of these side-effects. On the other hand, diabetes and hyperglycemia leads to increase in production of free radicals and oxidative stress [6]. Extremely high levels of free radicals lead to the biomolecular damages to membrane lipids, cellular proteins, nucleic acids, ultimately cell death and tissue injuries. However, this drug did not have anti-oxidant activity. Hence, discovery and development of new antidiabetic drugs from natural plant sources with high therapeutic potency, less adverse effects and anti-oxidant properties is at the center of intense research effort.
NAD is known as a central metabolic regulator and can help the mitochondrial competence. NAD is a fundamental compound for maintain the health of some tissues like nerves and heart. Unfortunately, aging has negative effect on NAD+ levels constantly via NADase called CD38. The levels of this enzyme increase during aging [7]. There are two intermediates of NAD+ and they are effective to increase the concentration of NAD+ in several tissues whose names are NR and NMN. In July 2013, the NR supplements were accessible with the brand name NIAGEN (Chromadex Incorporated, Irvine, California, USA) [8]. In addition, recent studies have indicated that NMN is capable of improving neuronal function such as memory and cognition in the brain of rat and mouse models of Alzheimer’s disease and can act as an inhibitor of neuron death after ischemia or intra cerebral hemorrhage [9]. Recent studies have reported that NMN is capable of improving mitochondrial function in several metabolic organs such as skeletal muscle, liver, heart and eyes [10]. Eventually, the long-term (1-year) oral administration of NMN (over 300 mg/kg) is harmless and apparent complications or toxic effects in normal wild-type C57BL/6 mice have not been observed [9]. Moreover, recent studies illustrate that supplements with NR protects against high-fatdiet (HFD)-induced obesity in mice [11]. Also, NMN can ameliorate and protect the hepatic function and diabetic neuropathy respectively. Therefore, these studies indicate that NMN and NR could offer broad applications and therapeutic potential.
Nucleosides are created by a nitrogenous base attached to a pentose sugar via a glycosidic linkage between the N-3 nitrogen of a purine or the N-1 nitrogen of a pyrimidine, and the C1’ carbon of the pentose sugar. A nucleoside plus a phosphate group yields a nucleotide. ADN, cytosine (CYT), INO and AMP are examples of nucleosides and nucleotides.
Nucleosides, NR, NMN and INO were identified and most of them exist in our daily foods quite differently, including vegetables, fish, meat, and fruits. In addition, recent studies have reported that human and cow milk contains these compounds at micromolar concentrations [12-15]. Also, these compounds are usually used as a dietary supplement to raise the daily dietary intake of nutrients. INO is mostly utilized by athletes to handle intense training or strenuous exercise with no fatigue in muscles. INO has another characteristic which has anti-inflammatory effects. INO inhibits the pro-inflammatory materials and protects tissues from inflammation induced by endotoxin and reperfusion injury [16]. Supplements of INO can help the CNS to preventing the second injury after spinal cord injury (SCI) [17]. The exact mechanisms of INO activity have still not recognized, but in some neurons it can intervene in gene expression of axon outgrowth [18]. In some studies the axonal rewiring in rats and nueroprotective effects of INO in vitro and in vivo has been documented [19].
Since the functional groups (OH group) of nucleosides, INO, NR, NMN, ADP-ribose, ADPglucose, cAMP and NAD (Fig. 1) are similar to ACR and miglitol as main synthetic drugs in inhibition of α-Amy/α-Glu, thus, it proposed that effects of inhibition of these enzymes by these compounds on glucose concentration can consider as one of the most important advantages of these compounds. Therefore, in this study the inhibitory activities of these compounds have been investigated against the α-Glu and α-Amy for their antihyperglycemia potential. This work will contribute towards the understanding of antihyperglycemia activity of nucleosides and their analogues, specifically towards the management and prevention of T2D.
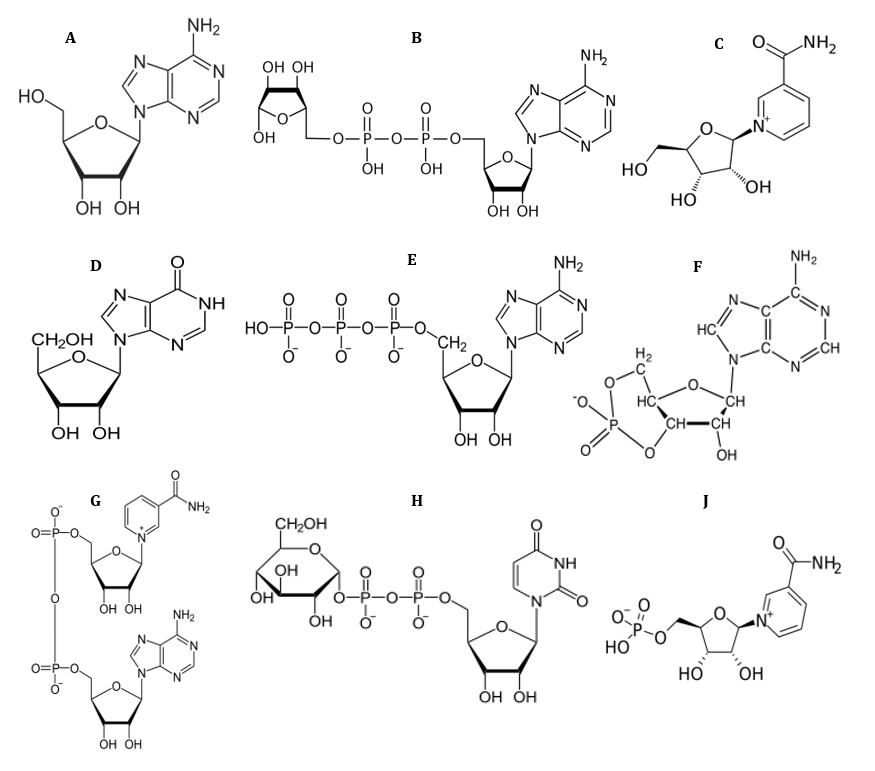
Fig 1. Chemical structure of ADN (A), ADP-ribose (B), NR (C), INO (D), ATP (E) and cAMP (F), NAD (G), ADPglucose (H) and NMN (J).
2.1. Chemicals
Our compounds, P-nitrophenyl alpha-d-glucopyranoside (p-NPG) and ACR were the antibiotics procured from the Sigma Aldrich Chemical Company. Ten mM of a sodium hydroxide solution was used to obtain the stock solutions, which were then diluted in a phosphate buffer.
2.2. Preparing rat intestinal acetone powder
A formerly-suggested technique was utilized with minor changes in the core process to gather intestinal acetone powder from the rats [20, 21]. The rats’ intestines were sectioned into smaller parts that were rinsed utilizing thirty ml of cold NaCl (0.9%). The temperature for the operations was 4 ◦C [22]. The intestines were homogenized using Potter–Elvehjem homogenizer in two vol. of five mM of phosphate buffer with pH 7.0. After an hour, ten vol. of the acetone solution cooled down to −20 ◦C was inserted into the homogenates. Shaking the mixture, it was then centrifuged for thirty minutes at 1500×g followed by ten minutes. The precipitates were also washed utilizing 12 vol. of cold acetone, then the mixture was centrifuged at 1500×g for thirty minutes. Overnight the precipitate was dried at 4 ◦C and kept at −20 ◦C prior to usage [22].
2.3. Rat α-Glu inhibition assay
On the basis of the method proposed by Yousefi, α-Glu activities were assayed following absorbance changes at 405 nm caused by p-NPG hydrolysis [20]. By dissolving the intestinal acetone powder in ice cold phosphate buffer (pH 7.0,10 mM), the α-Glu activity was measured, and the solution was sonicated at 4 ◦C for 25 s. The suspension was then centrifuged with 10,500×g for 75 min at 4 ◦C, and the resultant supernatant was dialyzed for 24 h against the same buffer. The reaction mixture including rat α-Glu and different concentrations of our compounds were pre-incubated at 37◦C for 10 min. The absorbance increase was then followed at 405 nm for thirty minutes. All the tests were repeated for at least 3 times.
2.4. α-Amy assay
α-Amy inhibitory activities related to the medicines were examined based on measuring glucose levels. α-Amy was incubated for ten minutes at 25 ◦C with the compounds and ACR. The test was carried out in 20 mM of a sodium phosphate buffer with a 6.9 pH. Then, after adding the starch solution (1% soluble starch), the reaction mixture was incubated for five minutes. The reaction was stopped followed by the addition of four hundred μl of the DNS reagent and boiling in a water bath for five minutes. After incubation of the solution and maintaining it at room temperature for fifteen minutes, 5 ml of water was introduced. By measuring the absorbance of the samples at 540 nm [23], it was compared with the experiment control absorbance. The findings were regarded as the criterion for the α-Amy inhibition percentage. The tests were repeated for three times.
2.5. Determining the inhibitory constant and inhibition fashion
To calculate the inhibitory constant and determine the inhibition mode, α-Glu activities were assayed at different concentrations of p-NPG in the occurrence of an increasing concentration of NAD, i.e. 0, 3, 6 and 12 µM. Moreover, α-Amy activities were assessed in various concentrations of starch (as substrate) by increasing the concentration of NAD, i.e. 0, 3, 6 and 12 µM, for determining the inhibition fashion and the inhibitory constant. The primary velocity, V0, was also calculated as the variations’ slope in absorbance at 540 nm in the catalytic reaction linear phase. For the competitive inhibition, the Lineweaver-Burk equation is presented as [24]:

and the secondary plot is made as:

where [I] indicates the inhibitor concentration, [S] is the substrate concentration, the apparent Michaelis-Menten constant, Ki shows the inhibition constant and Km represents the Michaelis-Menten constant, V is the enzyme reaction rate by existence and nonexistence of the inhibitors [24].
2.8. Molecular docking
2.8.1. Homology modeling and model verifying
The protein sequence and homology for Baker’s yeast a-Glu (MAL12) were modeled using the technique provided by Imran [25, 26]. Using SWISSMODEL a search that was conducted and proteins were identified in the Protein Data Bank (PDB) with high sequence similarity. The 72%-identical α-D glucose bound isomaltase with a similarity of 85% related to Saccharomyces cerevisiae (PDB ID: 3A4A) was exposed to homology modeling and sequence alignment using the automated homology modeling pipeline SWISS-MODEL controlled by the Swiss Institute of Bioinformatics [27]. PROCHECK was utilized to verify the quality of the established homology model.
2.8.2. Docking investigations
Docking studies were performed through the homology model of α-Glu and the α-Amy’s crystal structure with PDB ID:1PIF [28]. Based on Fig 1, the compounds’ molecular structure depicted via ACD/LAB was traced by changing it to the three-dimensional conformation utilizing Avogadro 1.2.0. For minimizing energy and optimizing ultimate geometry, the steepest-descend algorithm in Avogadro was utilized. Adding Gastiger charges and activating the rotational bonds through the MGL tools package, the drugs were docked into α-Amy/α-Glu via Auto Dock Vina. A box with 30*30*30-nm in the directions of X, Y, and Z were chosen in the search space by adjusting its center on the proteins’ active sites.
2.9. MD simulations
The ligand-protein’s complex structure with the least energy determined by the docking process was utilized as the initial geometry for the MD procedure. Gromos53a6 force-field, GROMACS toolkit-5.1 (www.gromacs.org) was used to perform the MD simulation [29]. NAD’s topology parameters and topology file were attained from the PRODRG server, and the charges were reallocated through the data related to the similar atom kinds in gromos 53a6 fore field [30]. The, the complexes were introduced into a periodic cubic box with 11×11×11-nm dimensions and a prolonged simple point charge (SPC/E) model of water utilized for dissolution [31]. Calcium (6 for α-Amy-NAD system) and sodium ions (2 for free α-Amy, 8 for α-Glu-NAD systems and 16 for free α-Glu) and were sufficiently added to the system for neutralizing. Within the system, 3D periodic boundary circumstances were also utilized. The energy was minimized through the steepest-descent technique for releasing undesired contacts in all the MD simulations, after 100 ps equilibration in NVT and NPT ensembles [32, 33]. A constant temperature of 300 K was obtained using velocity-rescale thermostat, and a constant pressure of 1 bar was attained by a Parrinello–Rahman barostat [34]. All the bonds were limited using he LINCS algorithm [35]. The long-range interactions were investigated using a 10 Å cut-off distance and the particle mesh Ewald technique [36]. The Verlet cut off scheme was utilized for a neighbour search, while updating the adjacent lists every 10 stages [37]. Ultimately, 50-ns MD simulations were performed using the leapfrog algorithm.
3.1. Determining the α-Glu/α-Amy inhibition potency based on IC50 values
The half maximal inhibitory concentration (IC50) value is a prevalent method due to measuring the inhibitor strength. As illustrated in Table. 1, the α-Glu activity was decreased in the presence of all compounds in a dose-dependent manner. The median inhibition concentration (IC50) values for α-Glu in the presence of ADN, AMP, NR, INO, ATP, NAD, ADPribose, ADP-glucose and NMN were determined 208.6±3.8, 254.1±5.2, 177.7±4.8, 192.1±5.2, 215.9±2.7, 55.4±1.3, 63.4±2.2, 75.6±4.2 and 196.1±2.6, respectively (Table. 1). Also, α-Amy is the main enzyme for breakdown of starch to more simple sugars (dextrin, maltotriose, maltose and glucose). α-Amy inhibitors play a fundamental role as the obstacles in absorption of dietary starch; therefore, they are well known as starch blockers. Starch is a complicated carbohydrate which firstly requires digestive enzymes including amylase in order to breaking down and then secondary enzymes for absorption. Hence, in this study starch was used as the substrate of α-Amy enzyme. Our results indicate that all compounds are capable of inhibiting α-Amy activity in a dose-dependent manner. The IC50 values in the presence of ADN, AMP, NR, INO, ATP, NAD, ADP-ribose, ADP-glucose and NMN were determined 145.3±2.4, 202.3±3.9, 127.7±4.8, 163.5±3.6, 185.3±1.2, 80.4±2.8, 64.8±4.7, 51.1±1.6 and 166.5±1.4, respectively (Table. 1).
Table 1. IC50 values for α-Glu and α-Amy in the presence of different concentrations of compounds. Production of glucose was monitored by absorbance at 405 nm and 540 nm for α-Glu and α-Amy, respectively.
|
Compounds |
IC50 values for α-Glu (µM) |
IC50 values for α-Amy (µM) |
|
AD |
>1000 |
>1000 |
|
ADN |
208.6±3.8 |
145.3±2.4 |
|
AMP |
254.1±5.2 |
202.3±3.9 |
|
ATP |
215.9±2.7 |
185.3±1.2 |
|
INO |
192.1±5.2 |
163.5±3.6 |
|
NR |
177.7±4.8 |
127.7±4.8 |
|
NMN |
196.1±2.6 |
166.5±1.4 |
|
c-AMP |
>1000 |
>1000 |
|
NAD |
55.4±1.3 |
80.4±2.8 |
|
ADP-ribose |
63.4±2.2 |
64.8±4.7 |
|
ADP-glucose |
75.6±4.2 |
51.1±1.6 |
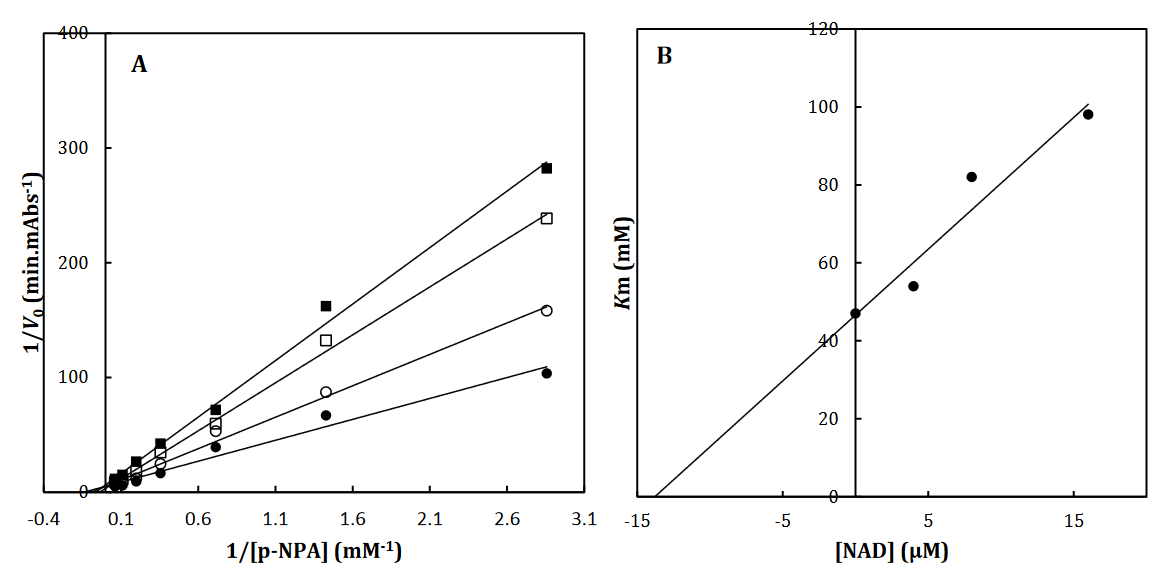
Fig 2. The Lineweaver-Burk plot of α-Glu in the absence (●) and the presence of 3 (○), 6 () and 12 (□) μM of NAD (A). The value of the inhibitory constant was determined from the negative value of the x-intercept of the plot. Secondary plot for inhibition of α-Glu with NAD (B). Data shown are representative example of three independent experiments and standard deviations were approximately within 5% of the experimental values.
3.2. Determining the inhibition mode and inhibition constant, Ki
To recognize the kinetic parameters and describe the inhibition activity of NAD, Kinetic assessments were performed utilizing the Lineweaver–Burk double reciprocal plot. A straight line is mainly yielded by the plot of 1/V versus 1/[S] with an intercept of 1/Vmax and a slope of Km/Vmax. A pattern of line is obtained by overlapping the double reciprocal lines for an enzyme reaction at various constant concentrations of inhibitor as the feature of a specific kind of inhibitor. Ki is determined using the graph of the of the Lineweaver– Burk plot’s slope against [I], namely the secondary plot, which is attained from the plot’s xintercept. Based on the Lineweaver–Burk plot (Fig. 2A), it is possible to explain the α-Glu inhibition kinetics of NAD by the competitive-state inhibition. In general, the competitive inhibitions are classified into 3 kinds of hyperbolic, linear, and parabolic, which can be recognized by their secondary plots’ shape. Considering the linear form of both the primary (Lineweaver–Burk) and secondary plots of the α-Glu inhibition by NAD (Fig. 2A-B), the observed inhibition can be linear competitive. Thus, the substrate (p-NPG) and inhibitor (NAD) are capable of competing for binding to the same active site. Km was plotted versus different concentrations of the drugs (secondary plot) (Fig. 2B) to determine the Ki of NAD as 4.4± 0.6 µM. The inhibition mode and constant of α-Amy were determined in the present study by existing NAD. The α-Amy inhibition kinetics of NAD was described well by the mixed-state inhibition based on Fig. 3A and the Lineweaver–Burk plot. Drawing the secondary plot yields the Ki of NAD as 8.4±0.8 µM in the low micro molar range (Fig 3).
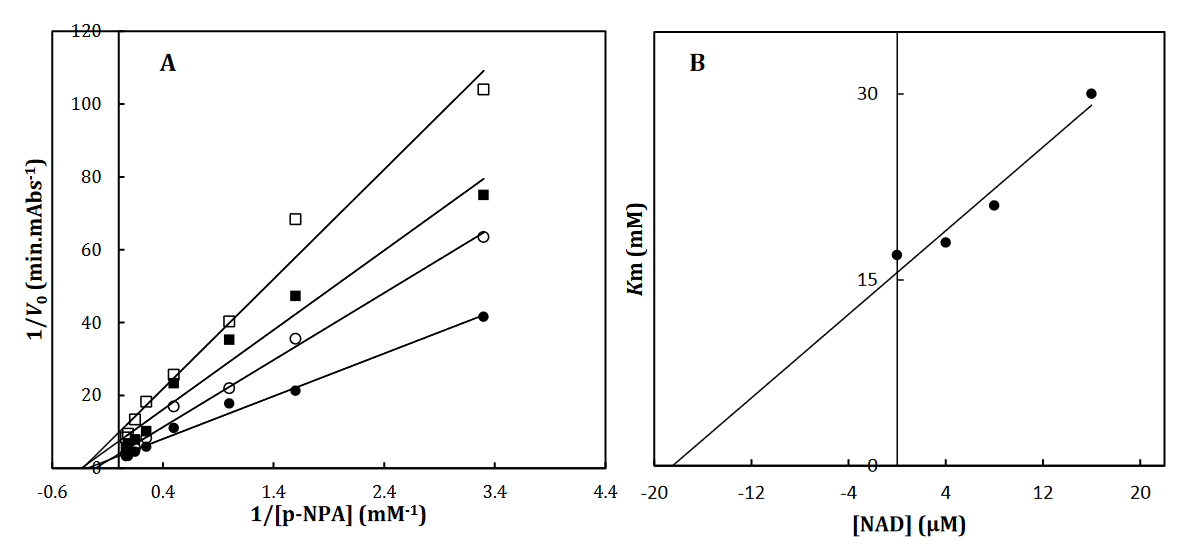
Fig 3. The Lineweaver-Burk plot of α-Amy in the absence (●) and the presence of 3(○), 6 () and 12 (□) μM of NAD (A). The value of the inhibitory constant was determined from the negative value of the x-intercept of the plot. Secondary plot for inhibition of α-Amy with NAD (B). Data shown are representative example of three independent experiments and standard deviations were approximately within 5% of the experimental values.
3.4. Molecular simulation and docking analysis
To better comprehend the enzyme-inhibitory interactions mechanism, the molecular docking method was employed. This technique was also utilized for predicting the location of the drugs in the α-Amy/α-Glu active site. The In-silico docking indicated that tentatively decided hindrance data were accompanied to the docking outcomes supporting the legitimacy of the used in-silico methods. Since NAD provided the least binding energies while interacting with α-Glu and α-Amy, it was considered to conduct the molecular dynamic simulation in subsequent phase. Performing the MD simulations, the amino acids were investigated included in the binding of NAD to α-Amy/α-Glu, and more exact models were obtained for the ligand interaction with these enzymes in fairly actual/natural circumstances.
NR, INO, ATP, NAD, ADP-ribose, ADP-glucose, cAMP, NMN and ACR as a commercial antidiabetic drug showed binding energies of -6.8, -6.9, -7.3, -8.5 -7.4, -7.6, -6.3, -7.0 and -7.4 kcal/mol, to combined with active site of α-Amy, respectively. Fig. 7 represents the graphical cooperation between the drugs and the α-Amy’s active site, and their connecting potential to the α-Amy’s nucleophilic active site. Based on the virtual docking results, D300, H305, D197, Y62, W59, Y151,Q63, Y163, L165, H201, W59, R195, H299 and I235 residues were found as the robust amino acids for interacting with the drugs. Moreover, the interaction elements’ dynamics was examined through MD simulations. The framework equilibration and basic solidness are normally evaluated through the root-mean-square deviation (RMSD). In Fig. 5A, the plots of the RMSD values are represented for α-Amy alone and the protein spine in the α-Amy-NAD complex between 0 and 50 ns. As observed in Fig. 5A, after 25000 ps of the simulation, the RMSD values of α-Amy-NAD frameworks and α-Amy alone achieved a relative equilibration, then they fluctuated nearby the normal values. Based on this compelling evidence, the reproduction time adequacy and the system reaching the relentless state are suggested. Based on Fig. 5B, by determining the RMSF values for the αAmy-NAD complex and α -Amy alone, the alterations in the protein residual fluctuations surrounding their normal qualities were analyzed. The findings indicated minor variations in most of the residues of α-Amy in the α-Amy-NAD complex, indicating the incidence of a stable connotation. The obtained Rg values utilizing MD simulation demonstrate that the Rg values of α-Amy alone do not demonstrate a huge difference with the values for the αAmy-NAD frameworks. The Rg values of α-Amy alone and α-Amy-NAD demonstrate some compactness of protein in the complex framework. The changes of protein residual fluctuation around their normal qualities were scrutinized by computing RMSF values for α-Amy alone and α-Amy-NAD complex as displayed in Fig. 5C. The results indicated minor fluctuations in most residues of α-Amy in the complex with NAD suggesting that stable association has occurred. Similarly, solvent accessible surface area (SASA) was assessed with the results (Fig. 5D) revealing reduction in both systems. The extent of reduction in SASA levels of protein-NAD system was slightly lower than in free protein. Considering the Rg data, these results can be another sign for protein compactness in the presence of drug.
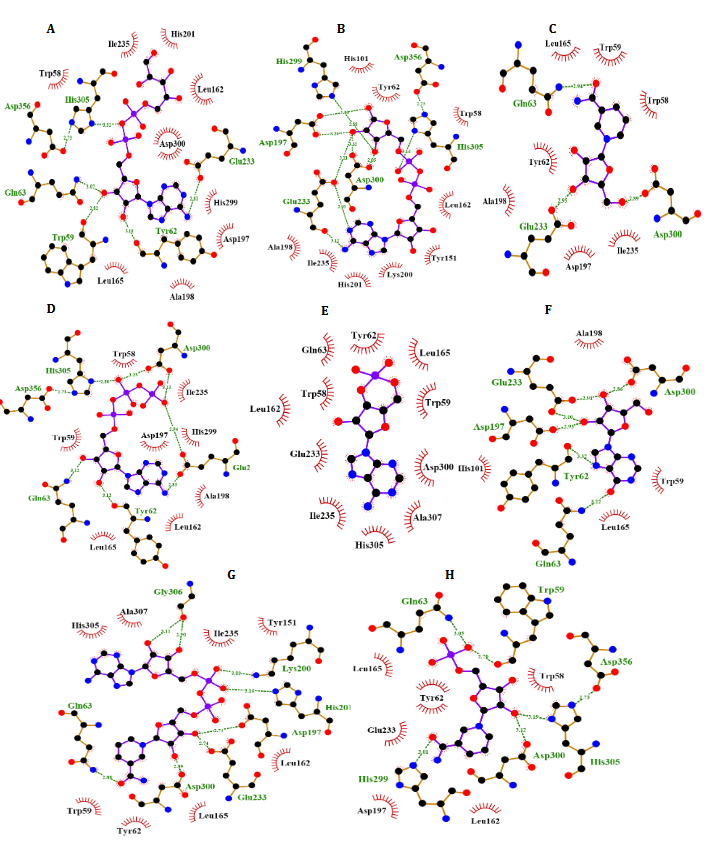
Fig. 4. Molecular docking of α-Amy. Two dimensional diagram shows interactions between binding site of αGlu and compounds (ADP-glucose (A), ADP-ribose (B), NR (C), ATP (D), Camp (E), INO (F), NAD (G), NMN (H)). (For interpretation of references to color in this figure legend, the reader is referred to the web version of this article).
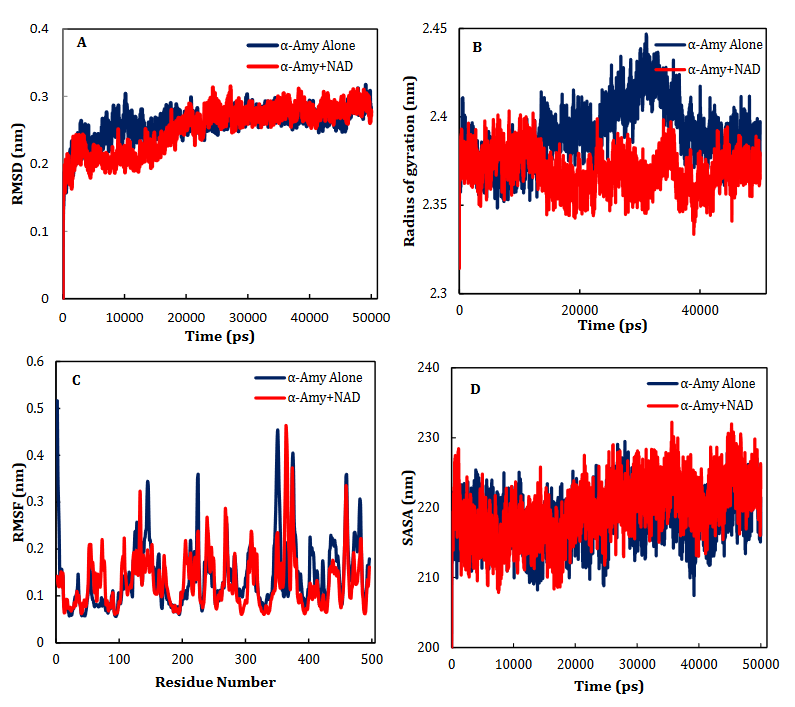
Fig. 5. Time dependencies of the RMSDs (nm) of the α-Amy alone (blue) and α-Amy-NAD (red) systems (A). Time dependencies of radius of gyration from starting structure for the α-Amy alone (blue) and α-Amy-NAD (red) systems in the 50-ns MD simulation (B). The RMSF and SASA of the α-Amy alone (blue) and α-Amy-NAD (red) systems (C-D).
α-Glu is another digestive carbohydrate enzyme playing a vital (pivotal) role in development of diabetes. By performing docking and molecular dynamic simulations, the experimental results were verified to identify amino acids in the α-Glu active site included in communication with the inhibitors (the medicines and AC) and to attain data regarding the components of the complex framework. Considering the inaccessibility of the PDB structure of α-Glu for eukaryotic organisms including rats and humans, α-Glu of S.cerevisiae was attained utilizing the homology modeling. Based on Fig. 9, N241, H439, D408, F157, K155, S15, H239, F177, H279, R312, N412, S384, P309, and R439 residues seem to be essential amino acids in interactions with these drugs. The residues have a significant role in maintaining the inhibitors, however, they bind with the enzyme’s active site. There is a complete consistency between the existing docking and findings for AC entirely and the detailed results previously gained by the authors. NR, INO, ATP, NAD, ADP-ribose, ADPglucose, cAMP, NMN and ACR (as a commercial anti-diabetic drug) have binding affinity energies of -7.8, -7.3, -8.3, -10.0, -8.7, -8.9, -6.6, -6.8, and -7.9 (kcal/mol) to interact with active site of α-Glu, respectively. Based on Fig. 7A, for the molecular elements, the RMSD values of α-Glu-NAD frameworks and α-Glu alone reached equilibration followed by 32000 ps of the reenactment time flocculate around the normal value. To verify the protein compactness in the complex with NAD, the RMSF findings (Fig. 7.B) can be obtained. The results of RMSF and SASA (Fig. 7C–D) with the Rg and RMSD data can be considered to prove the protein compactness in the complex with the drug.
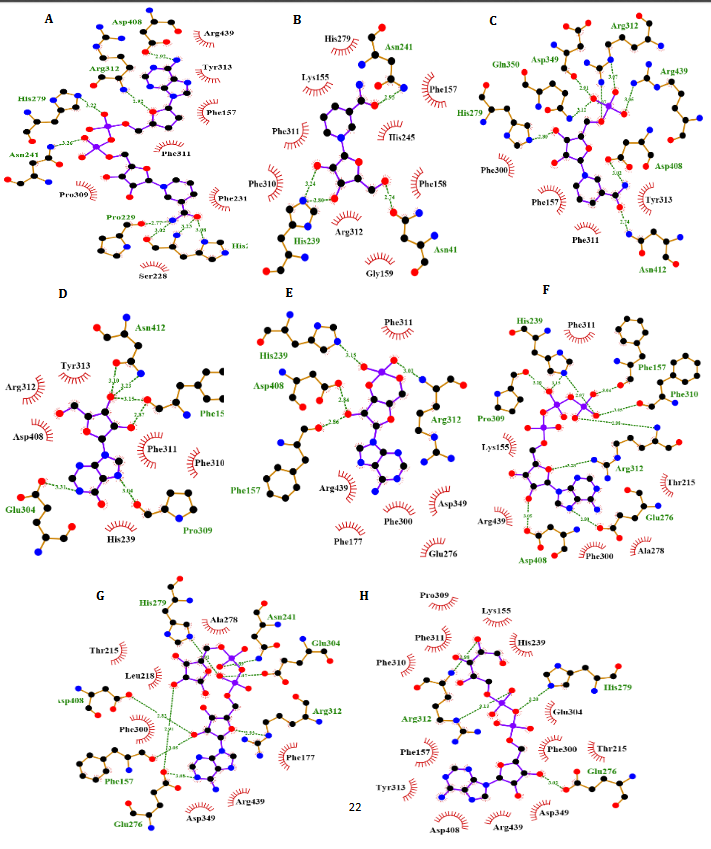
Fig. 6. Molecular docking of α-Glu. Two dimensional diagram shows interactions between binding site of αGlu and compounds (NAD (A), NR (B), NMN (C), INO (D), cAMP (E), ATP (F), ADP-ribose (G), ADP-glucose (H)). (For interpretation of references to color in this figure legend, the reader is referred to the web version of this article).
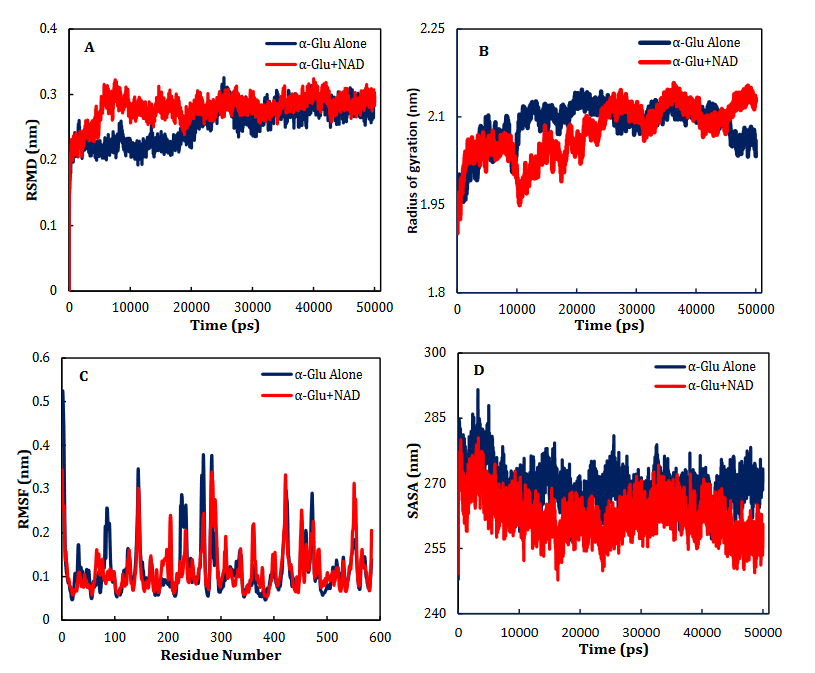
Fig. 7. Time dependencies of the RMSDs (nm) of the α-Glu alone (blue) and α- Glu-NAD (red) systems (A). Time dependencies of radius of gyration from starting structure for the α-Glu alone (blue) and α- Glu-NAD (red) systems in the 50-ns MD simulation (B). The RMSF and SASA of the α-Glu alone (blue) and α- Glu-NAD (red) systems (C-D).
4.1. Hyperglycemia
Hyperglycemia is an abnormal increase in blood glucose level and an etiology of T2D; a disease triggered by insulin resistance [38, 39]. According to numerous studies, a key treatment of T2D is to reduce the post-prandial hyperglycemia by delaying the absorption of glucose via inhibiting the enzymes, α-Glu and α-Amy, in the digestive tract [40-42]. The glucose absorption rate is delayed by enzyme inhibitors by prevention of carbohydrate digestion and accordingly dampening the increase in the postprandial plasma glucose. The starch digestive enzymes inhibition by synthetic agents like ACR, is a key clinical approach to control the postprandial glycemia [43-46]. Though ACR , which is confirmed by the Food and Drug Administration, decreases blood glucose levels, it is reported that this inhibitor causes critical side effects like abdominal distention, flatulence, diarrhoea and pneumatosis cystoides intestinalis and the lack suitable antioxidant activity, while enzyme inhibitors with plant basis are potentially safer [47]. Therefore, it is an urgent task to search for natural and non-toxic α-Amy/α-Glu inhibitors without any adverse or unwanted secondary effects, high therapeutic potency and suitable anti-oxidant activity.
According to enzymatic and chemical assessments, enzymatic hydrolysis by α-Amy results in cleave the linkage within the two glucose units [48]. It is extensively identified that the existence of two carboxyl-containing amino acids is required for the two-step mechanism to retain glycoside hydrolases. One acts as an acid/base catalyst and the other has the role of a nucleophile in charge of creating the glycosyl-enzyme intermediate [49]. Numerous works reported that both Glu233 and Asp197 may be needed for producing the â-linked glycosyl-enzyme intermediate. McCarter and Withers confirmed that Asp197 has the role of the catalytic nucleophile. As stated in former study, Glu233 that is very close to the chloride ion, should act as the catalytic acid/base [50]. It was reported that α-Amy inhibition by flavonoids strongly depends on the position and nature of the substituents. For instance, it was shown that the existence of –OH groups at different locations and the C=C double bond have different inhibitory impacts [49, 51]. As mentioned, RF has various functional groups like NH2, OH, N, NH, CO2, C=O and C=C, capable of interacting with the active site of enzymes utilizing hydrogen bond donors/acceptors. Therefore, since RF has anti-oxidant activity and the flexible backbone, numerous existing hydrogen bond donors/acceptors and aromatic rings well-suited structural characteristic for inhibition of α-Amy/α-Glu enzymes, in this study we investigated anti-diabetic effects NAD. In addition, though the synthesis and discovery of innovative α-Amy/α-Glu inhibitors are vital in their own right, it is similarly vital to assess their particular mechanism of inhibition versus enzymes included in carbohydrate digestion. Therefore, in this study not only we investigated effects NAD against α-Amy/α-Glu activity, but also we selected different analogs with at least an identical part for finding the most important parts in inhibition these enzymes. They differ both in their location of attachment and number of these functional groups and in their rings.
As shown in Table. 1, adenine inhibited α-Amy and α-Glu activity in concentrations more than 1 mM, while α-Amy and α-Glu activity suppressed in the presence of ADN with IC50 less than 200 µM. Since, ribose molecule is only different between adenine and ADN, thus, with comparing IC50 of adenine and ADN against α-Amy and α-Glu activity we found that ribose molecule plays the most important role in inhibition these enzymes. Also, we selected AMP that has a phosphate group attached to C5 ribose ring for comparing with ADN. Our results showed that ADN inhibited α-Amy and α-Glu activity with IC50 less than AMP. Also, similar results were observed in other analogs between NMN and NR. Therefore, we can conclude that the minor difference in the activity of these analogs can be due to the difference functional groups on the ribose part so that we can concluded that phosphate groups neither have impact or less impact on α-Amy/α-Glu inhibitory in comparison with hydroxyl groups. In addition, cAMP inhibited α-Amy and α-Glu activity with IC50 weaker than ADN and AMP. As shown in Fig. 1, cAMP has only one hydroxyl group and a rigid structure with limit torsions at ribose ring. Therefore, it is indicated that the lack of presence of hydroxyl groups at C2 and C5 positions on the ribose ring have weakened the affinity. On the other hand, for investigating the importance of hydroxyl groups and hydrogen acceptor/donor bonds we chosen compounds with more hydroxyl and amine groups. Accordingly, in this study we selected NAD, ADP-ribose and ADP-glucose that have at least four hydroxyl and one amine groups. As shown in Table. 1, NAD, ADPribose and ADP-glucose suppressed α-Amy and α-Glu activity with IC50 less than ADN, AMP and other compounds. Since, with increasing in number of hydroxyl and amine groups we observed an increase in α-Amy and α-Glu inhibitory, it was concluded that the number of hydroxyl and amine groups have an great impact on α-Amy/α-Glu inhibitory.
According to Fig. 4, our docking data implied that Glu233, His299, Asp356, Phe144, Asp197, Gln63, Asp300, Phe282, His305, Tyr62 and Trp59 play vital roles at active site of α-Amy interacting with these compounds. Furthermore, Asp408, Phe157, Phe177, Asn241, Arg439, Lys155, His239, Arg312, His245, Phe300, and His279 exists at the entry of the pocket and into the active site of α-Glu that are positively charged at experimental pH 7.2 and interacted with the hydroxyl groups of the compounds. Moreover, aspartate, asparagine, phenylalanine and tryptophan amino acids like Asn412, Glu276, Phe158, Phe310, Phe313, and Trp430 at active site, are both electron acceptors and donors with negatively charged at natural pH. In compared to the docking and experimental results on α-Glu and α-Amy together indicated that α-Glu is inhibited by these compounds with less IC50 and more potency. Comparison of amino acids included at active sites of α-Glu and α-Amy implied that further Asp, Arg, Glu, His, Lys amino acids exist with positive charges at the α-Glu active site that may influence the better interactions with these compounds’ hydroxyl groups. Furthermore, it is normally supposed that carbohydrate mimics with nitrogen such as ACR and miglitol were protonated in the active site and function as α-Glu and α-Amy inhibitors due to their potential to imitate the shape and charge of the transition state supposed for the enzymatic glycoside hydrolysis [52, 53].
Accordingly, since in previous studies it has been demonstrated that oral administration of NMN, NR and INO are safe even in high doses and also they exhibited a wide variety of biological activities like anti-inflammatory, antioxidant, thus, they can be suggested as new α-Amy and α-Glu inhibitors.
4.2. Hypoglycemia
So far in this article, we have discussed the advantages of these compounds, but we must explain about lack using of these compounds because they may potentiate the hypoglycemia effects of non-diabetic and anti-diabetic drugs. Today, numerous medicines are utilized in different ways to reduce and control blood glucose levels thus treating T1D and T2D. Most of these drugs are able to provide hypoglycemia by interfering with the glucose metabolism, decreasing the insulin clearance and stimulation of the insulin release[54]. From a medical viewpoint, hypoglycemia denotes for a serious condition causing the body not to be able to meet its energy needs. In case dramatic drop in blood glucose levels, there won’t be adequate energy for the body to properly function. Hypoglycemia can impose a danger for normal functionality of the brain, considering the dependence of the brain on the circulating glucose as its main energy source [55]. Severe hypoglycemia is associated with the incremented morbidity and even mortality in some cases in T1D as well as T2D. Since T2D is more usual than T1D by a factor of 10, severe hypoglycemia in patients with T2D is more problematic clinically [56].
Drugs are regarded as the main cause of hypoglycemia, particularly in diabetics [57]. Even in carefully management of diabetes, the utilized drugs can lead to medication-induced low blood sugar [58]. Furthermore, this condition is established in non-diabetics after taking the medicines for treatment of diabetes [59]. Though hypoglycemia is a well-defined adverse impact of anti-diabetic agents, various groups of pharmaceutical non-diabetic agents utilized daily in clinics can also contribute to the severe hypoglycemia. In Table 2, the absolute risk of emerging hypoglycemia related to commonly-utilized glucose-reducing agents for diabetes management is provided [54, 60]. In definite cases, non-diabetesrelated drugs lead to the less blood sugar (Table 3) [61-63].
|
Table 2. Risk of Hypoglycemia With Commonly Used Glucose-Lowering Agents [54] |
||
|
Drug Class |
Drug |
Mechanism of Glucose-Lowering Effects |
|
α-Glucosidase inhibitors |
Acarbose Miglitol |
Delay the process of digestion and absorption of carbohydrates in the small intestine |
|
Amylinomimetic |
Pramlintide |
Acts centrally to slow gastric emptying, suppress postprandial glucagon secretion, and decrease food intake |
|
Biguanides |
Metformin |
Decreases hepatic glucose production and intestinal absorption of glucose, improves insulin sensitivity of cells |
|
DPP-4 inhibitors |
Sitagliptin Saxagliptin |
Inhibit glucagon release
|
|
sulfonylureas |
Glimepiride Glipizide Glyburide |
Increase insulin release from the beta cells in the pancreas
|
|
Meglitinides |
Nateglinide Repaglinide |
Help the insulin-producing beta cells in the pancreas to release insulin |
|
Glucagon-like peptide-1 |
Exenatide Liraglutide |
Encourages the release of insulin from the pancreas, and holds back glucagon release |
|
Thiazolidinediones |
Pioglitazone Rosiglitazone |
Improve the patients sensitivity to insulin by reducing circulating fatty acid concentrations and lipid availability in liver and muscle |
|
Table 2. Non-Diabetes Drugs Associated with Hypoglycemia [54] |
||
|
Drug Class |
Drug |
Mechanism of Glucose-Lowering Effects |
|
ACE inhibitors |
Benazepril Enalapril Lisinopril Perindopril Ramipril Captopril Fosinopril Moexipril Quinapril Trandolapril |
Indirectly increases insulin sensitivity by increasing circulating kinins, which leads to vasodilatation in the muscles and increased glucoseuptake in muscle tissue
|
|
β-Blockers |
Noncardioselective: Levobunolol Metipranolol Nadolol Propranolol Sotalol Timolol
Cardioselective: Acebutolol Atenolol Betaxolol Bisoprolol Esmolol Nebivolol Metoprolol |
Inhibits glycogenolysis; attenuates signs and symptoms
|
|
Chloramphenicol |
|
May inhibit the metabolism of SUs |
|
Chloroquine |
|
Unknown |
|
Clofibrate |
|
Enhances the effect of SUs |
|
Disopyramide |
|
Unknown; appears to result from endogenous insulin secretion |
|
Ethanol |
|
Impairs gluconeogenesis; increases insulin secretion |
On the other hand, there is a disorder with many severe complications which is critical, complex, and extremely debilitating chronic illness called chronic fatigue syndrome (CFS). Its cause, diagnostic test, and effective treatment have not recognized yet. CFS is known with prolonged, debilitating, and relapsing fatigue and these patients suffer from several symptoms bringing about significant disability for at least around a half year and regularly for a considerable length of time [64]. In recent studies NADH and CoQ10 deficiencies are depicted in fibromyalgia (FMS) and CFS patients [65]. Both NADH and CoQ10 have a fundamental role in producing mitochondrial ATP and cellular metabolism homeostasis
[65]. Therefore, both CoQ10 and NADH can be considered as potential drugs to remedy the CFS and perhaps other chronic fatiguing illnesses. In some cases, patients are located in compulsory situation of multi-pharmacological therapeutic regimens with numerous drugs. Accordingly, since in this study have found that NADH can inhibit α-Amy and α-Glu at micromolar concentrations, thus consumption this drug with diabetic and non-diabetics drugs that can lead to hypoglycemia should be reconsidered by specialist.
Moreover, the risk of stroke is predictable in diabetic patients with 1.5 times more than people who do not suffer from diabetes. Citicoline or cytidine diphosphate-choline (CDPcholine) is an endogenous intermediate which play a key role in biosynthesis of phosphocholine. In addition, it is used as a drug to treat stroke and brain injury [66]. This intermediate is capable of entering the blood circulation and crossing the blood-brain barrier with converting to acetylcholine and phosphocholine. Also, citicoline used for
Alzheimer's disease and other types of dementia, head trauma, memory loss related to age, Parkinson's disease, attention deficit-hyperactive disorder (ADHD), and glaucoma [67].
Because of inhibitory characteristic of cytidine for α-Amy and α-Glu and locating patients in compulsory condition of multi-pharmacological therapeutic procedures, prescribing citicoline along with diabetic and even non-diabetic drugs which cause hypoglycemia should be reconsidered by specialist.
In this work, we assessed the interactions between some ADN, AMP, NR, INO, ATP, NAD, ADP-ribose, ADP-glucose, cAMP and NMN with α-Amy/α-Glu enzymes in-vitro. According to the findings, these compounds can be considered as α-Amy/α-Glu inhibitors. Comparing IC50 and Ki values of these compounds with ACR in previous studies (as a commercial antidiabetic drug), it was understood that these compounds can bind to the active sites of αAmy and α-Glu enzymes almost slightly weaker than ACR could through competitive-mode and mixed-mode inhibition, respectively. Furthermore, the experimental observations were confirmed by the docking studies. Thus, these hydrolyzing enzymes inhibitions can be considered a benefit of these compounds (or a side effect). Furthermore, as stated earlier considering the fact that insulin-induced hypoglycemia may take place in DM and other diseases, therefore, it is necessary to take caution in administration of these compounds under certain hypoglycemic circumstances for patients and to reconsider by specialists.
H. King, R.E. Aubert, W.H. Herman, Diabetes care, 21 (1998) 1414-1431. PMid:9727886
View Article PubMed/NCBID. Kalita, D.G. Holm, D.V. LaBarbera, J.M. Petrash, S.S. Jayanty, PLoS One, 13 (2018) e0191025. PMid:29370193
View Article PubMed/NCBIH.E. Lebovitz, The American journal of cardiology, 88 (2001) 20-25. 01833-1
View ArticleC.M. Sena, C.F. Bento, P. Pereira, F. Marques, R. Seiça, in: New Strategies to Advance Pre/Diabetes Care: Integrative Approach by PPPM, Springer, 2013, pp. 29-87.
View ArticleA. Hisamoto, T. Mizushima, K. Sato, Y. Haruta, Y. Tanimoto, M. Tanimoto, K. Matsuo, Internal Medicine, 45 (2006) 73-76. PMid:16484742
View Article PubMed/NCBIF. Giacco, M. Brownlee, Circulation research, 107 (2010) 1058-1070. PMid:21030723
View Article PubMed/NCBIE. Verdin, Science, 350 (2015) 1208-1213. PMid:26785480
View Article PubMed/NCBIK.L. Bogan, C. Brenner, Annu. Rev. Nutr., 28 (2008) 115-130. PMid:18429699
View Article PubMed/NCBIK.F. Mills, S. Yoshida, L.R. Stein, A. Grozio, S. Kubota, Y. Sasaki, P. Redpath, M.E. Migaud, R.S. Apte, K. Uchida, Cell metabolism, 24 (2016) 795-806. PMid:28068222
View Article PubMed/NCBIJ.B. Lin, S. Kubota, N. Ban, M. Yoshida, A. Santeford, A. Sene, R. Nakamura, N. Zapata, M. Kubota, K. Tsubota, Cell reports, 17 (2016) 69-85. PMid:27681422
View Article PubMed/NCBIC. Cantó, R.H. Houtkooper, E. Pirinen, D.Y. Youn, M.H. Oosterveer, Y. Cen, P.J. Fernandez-Marcos, H. Yamamoto, P.A. Andreux, P. Cettour-Rose, Cell metabolism, 15 (2012) 838-847. PMid:22682224
View Article PubMed/NCBIP. Bieganowski, C. Brenner, Cell, 117 (2004) 495-502. 00416-7
View ArticleS. Ummarino, M. Mozzon, F. Zamporlini, A. Amici, F. Mazzola, G. Orsomando, S. Ruggieri, N. Raffaelli, Food chemistry, 221 (2017) 161-168. PMid:27979136
View Article PubMed/NCBIS.A. Trammell, L. Yu, P. Redpath, M.E. Migaud, C. Brenner, The Journal of nutrition, 146 (2016) 957-963. PMid:27052539
View Article PubMed/NCBIH. Liu, Y. Jiang, Y. Luo, W. Jiang, Food Technology & Biotechnology, 44 (2006).
A. Wakai, D.C. Winter, J.T. Street, R.G. O'Sullivan, J.H. Wang, H.P. Redmond, Journal of Surgical Research, 99 (2001) 311-315. PMid:11469903
View Article PubMed/NCBIM. Kuricova, V. Ledecky, T. Liptak, A. Madari, I. Grulova, L. Slovinska, M. Nagyova, D. Cizkova, Neurological Sciences, 35 (2014) 1785-1791. PMid:24913204
View Article PubMed/NCBIB. Petrausch, R. Tabibiazar, T. Roser, Y. Jing, D. Goldman, C.A. Stuermer, N. Irwin, L.I. Benowitz, Journal of Neuroscience, 20 (2000) 8031-8041. PMid:11050124
View Article PubMed/NCBIL. Zai, C. Ferrari, S. Subbaiah, L.A. Havton, G. Coppola, S. Strittmatter, N. Irwin, D. Geschwind, L.I. Benowitz, Journal of Neuroscience, 29 (2009) 8187-8197. PMid:19553458
View Article PubMed/NCBIR. Yousefi, M.-M. Alavian-Mehr, F. Mokhtari, F. Panahi, M.H. Mehraban, A. Khalafi-Nezhad, Journal of enzyme inhibition and medicinal chemistry, 28 (2013) 1228-1235. PMid:23043430
View Article PubMed/NCBIB.B. Aggarwal, Y.-J. Surh, S. Shishodia, The molecular targets and therapeutic uses of curcumin in health and disease, Springer Science & Business Media, 2007. PMid:17569205
View Article PubMed/NCBIF. Panahi, R. Yousefi, M.H. Mehraban, A. Khalafi-Nezhad, Carbohydrate research, 380 (2013) 81-91. PMid:23978663
View Article PubMed/NCBIP. Bernfeld, (1955).
Y. Wang, G. Zhang, J. Yan, D. Gong, Food chemistry, 163 (2014) 226-233. PMid:24912720
View Article PubMed/NCBIL. Sim, K. Jayakanthan, S. Mohan, R. Nasi, B.D. Johnston, B.M. Pinto, D.R. Rose, Biochemistry, 49 (2009) 443-451. PMid:20039683
View Article PubMed/NCBIS. Imran, M. Taha, N.H. Ismail, S.M. Kashif, F. Rahim, W. Jamil, M. Hariono, M. Yusuf, H. Wahab, European journal of medicinal chemistry, 105 (2015) 156-170. PMid:26491979
View Article PubMed/NCBIM. Biasini, S. Bienert, A. Waterhouse, K. Arnold, G. Studer, T. Schmidt, F. Kiefer, T.G. Cassarino, M. Bertoni, L. Bordoli, Nucleic acids research, 42 (2014) W252-W258. PMid:24782522
View Article PubMed/NCBIM. Machius, L. Vértesy, R. Huber, G. Wiegand, Journal of molecular biology, 260 (1996) 409421. PMid:8757803
View Article PubMed/NCBIM. Abraham, D. Van Der Spoel, E. Lindahl, B. Hess, GROMACS user manual version, 5 (2014).
D.M. Van Aalten, R. Bywater, J.B. Findlay, M. Hendlich, R.W. Hooft, G. Vriend, Journal of computer-aided molecular design, 10 (1996) 255-262. PMid:8808741
View Article PubMed/NCBIH.J. Berendsen, J.P. Postma, W.F. van Gunsteren, J. Hermans, in: Intermolecular forces, Springer, 1981, pp. 331-342.
View ArticleR. Fletcher, M.J. Powell, The computer journal, 6 (1963) 163-168.
View ArticleG. Bussi, D. Donadio, M. Parrinello, The Journal of chemical physics, 126 (2007) 014101. PMid:17212484
View Article PubMed/NCBIR. Martoň|k, A. Laio, M. Parrinello, Physical review letters, 90 (2003) 075503. PMid:12633242
View Article PubMed/NCBIB. Hess, H. Bekker, H.J. Berendsen, J.G. Fraaije, Journal of computational chemistry, 18 (1997) 1463-1472. 1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H
View ArticleT. Darden, D. York, L. Pedersen, The Journal of chemical physics, 98 (1993) 10089-10092.
View ArticleH. Grubmüller, H. Heller, A. Windemuth, K. Schulten, Molecular Simulation, 6 (1991) 121-142.
M.A. Gyamfi, M. Yonamine, Y. Aniya, General Pharmacology: The Vascular System, 32 (1999) 661-667. 00238-9
View ArticleJ. Kaur, Cardiology research and practice, 2014 (2014). PMid:24711954
View Article PubMed/NCBIJ.H. Kim, H.-J. Kim, M. Park, Applied Clay Science, 101 (2014) 272-276.
View ArticleW.D. Seo, J.H. Kim, J.E. Kang, H.W. Ryu, M.J. Curtis-Long, H.S. Lee, M.S. Yang, K.H. Park, Bioorganic & medicinal chemistry letters, 15 (2005) 5514-5516. PMid:16202584
View Article PubMed/NCBIA. Kato, N. Kato, E. Kano, I. Adachi, K. Ikeda, L. Yu, T. Okamoto, Y. Banba, H. Ouchi, H. Takahata, Journal of medicinal chemistry, 48 (2005) 2036-2044. PMid:15771446
View Article PubMed/NCBIE. Lo Piparo, H. Scheib, N. Frei, G. Williamson, M. Grigorov, C.J. Chou, Journal of medicinal chemistry, 51 (2008) 3555-3561. PMid:18507367
View Article PubMed/NCBIJ. Singh, A. Dartois, L. Kaur, Trends in Food Science & Technology, 21 (2010) 168-180.
View ArticleW.C. Obiro, T. Zhang, B. Jiang, British journal of nutrition, 100 (2008) 1-12. PMid:18331662
View Article PubMed/NCBIS. Lordan, T.J. Smyth, A. Soler-Vila, C. Stanton, R.P. Ross, Food chemistry, 141 (2013) 21702176. PMid:23870944
View Article PubMed/NCBIM. Ali Asgar, International Journal of Food Properties, 16 (2013) 91-103.
View ArticleH. Teng, L. Chen, T. Fang, B. Yuan, Q. Lin, Journal of functional foods, 28 (2017) 306-313.
View ArticleH. Teng, L. Chen, Critical reviews in food science and nutrition, 57 (2017) 3438-3448. PMid:26854322
View Article PubMed/NCBIJ.D. McCarter, G.S. Withers, Current opinion in structural biology, 4 (1994) 885-892. 90271-2
View ArticleL. Chen, H. Teng, Z. Xie, H. Cao, W.S. Cheang, K. Skalicka-Woniak, M.I. Georgiev, J. Xiao, Critical reviews in food science and nutrition, 58 (2018) 513-527. PMid:27438892
View Article PubMed/NCBIL. Chen, C. Gnanaraj, P. Arulselvan, H. El-Seedi, H. Teng, Trends in Food Science & Technology, 85 (2019) 149-162.
View ArticleL. Chen, X. Lu, H. El-Seedi, H. Teng, Trends in Food Science & Technology, 88 (2019) 46-56.
View ArticleM.H. Vue, S.M. Setter, Diabetes Spectrum, 24 (2011) 171-177.
View ArticleE. Bonora, R.A. DeFronzo, Diabetes Complications, Comorbidities and Related Disorders, Springer, 2018.
View ArticleS. Heller, P. Novodvorsky, Medicine, 47 (2019) 52-58.
View ArticleJ.-M. Guettier, P. Gorden, Endocrinology and Metabolism Clinics, 35 (2006) 753-766. PMid:17127144
View Article PubMed/NCBIJ.-F. Yale, B. Paty, P.A. Senior, Canadian journal of diabetes, 42 (2018) S104-S108. PMid:29650081
View Article PubMed/NCBIS. Kirk, M. Eckert-Norton, The Journal of Clinical Endocrinology & Metabolism, 98 (2013) 39A39A. PMid:24098022
View Article PubMed/NCBIS. Bolen, L. Feldman, J. Vassy, L. Wilson, H.-C. Yeh, S. Marinopoulos, C. Wiley, E. Selvin, R. Wilson, E.B. Bass, Annals of internal medicine, 147 (2007) 386-399. PMid:17638715
View Article PubMed/NCBIJ.T. DiPiro, R.L. Talbert, G.C. Yee, G.R. Matzke, B.G. Wells, L.M. Posey, Pharmacotherapy: a pathophysiologic approach, McGraw-Hill Education New York, 2014.
J.R. White Jr, R.K. Campbell, Endocrinology and metabolism clinics of North America, 29 (2000) 789-801. 70164-X
View ArticleJ.R. White JR, R. Campbell, The Diabetes Educator, 21 (1995) 283-289. PMid:7621729
View Article PubMed/NCBIJ. Castro‐Marrero, N. S|ez‐Franc{s, D. Santillo, J. Alegre, British journal of pharmacology, 174 (2017) 345-369. PMid:28052319
View Article PubMed/NCBIJ. Castro-Marrero, M.D. Cordero, M.J. Segundo, N. Sáez-Francas, N. Calvo, L. Román-Malo, L. Aliste, T. Fernandez de Sevilla, J. Alegre, in, Mary Ann Liebert, Inc. 140 Huguenot Street, 3rd Floor New Rochelle, NY 10801 USA, 2015.
R. Conant, A.G. Schauss, Alternative medicine review, 9 (2004) 17-31.
R. García-Cobos, A. Frank-García, M. Gutiérrez-Fernández, E. Díez-Tejedor, Journal of the neurological sciences, 299 (2010) 188-192. PMid:20875651
View Article PubMed/NCBI