María D. Guillén
Email: mariadolores.guillen@ehu.eus
© 2019 Sift Desk Journals. All Rights Reserved
VOLUME: 3 ISSUE: 1
Page No: 247-254
María D. Guillén
Email: mariadolores.guillen@ehu.eus
Bárbara Nieva-Echevarría, Encarnación Goicoechea, María D. Guillén*
Food Technology, Faculty of Pharmacy, Lascaray Research Center, University of the Basque Country (UPV/EHU).* Corresponding author: mariadolores.guillen@ehu.eus
Francesco Siano(francesco.siano@isa.cnr.it)
Meng Shi(11616052@zju.edu.cn)
Yasemin Bakircioglu Kurtulus(ybakircioglu@trakya.edu.tr)
B. Nieva-Echevarria, E.Goicoechea, M.D. Guill
During the last decade obtaining further knowledge on lipid digestion has become a challenging task in the field of Food Science and Nutrition research. However, the great complexity of this process requires the use of sound, accurate and simple analytical techniques which are able to provide as much information as possible; only thus can a global view and therefore a better understanding of the ongoing process be obtained. This review tackles the advantages and drawbacks of the different methodologies currently employed for this purpose, focusing on a new approach, recently developed and based on Proton Nuclear Magnetic Resonance (1H NMR) spectroscopy. This new methodological approach not only provides a great deal of information in a simple, rapid and accurate way, but also overcomes many of the disadvantages of the techniques employed to date. In this sense, 1H NMR can be considered a very promising alternative for research on lipid digestion, contributing to shed more light on the complex digestion process of lipids and the factors that may affect it.
Keywords: lipolysis, digestion, NMR spectroscopy, pH-stat titration, chromatography.
Consumer demand for healthier foods is a general trend, especially in Western countries where concern for maintaining and/or improving health status through diet has grown considerably. Nevertheless, the design of healthier foods requires, among other things, deeper knowledge of the food digestion process and of the fate of the different macro and micronutrients in the gastrointestinal tract until their absorption into the bloodstream. In this context, special attention is nowadays being paid to the study of food lipids and the digestion process to which they are submitted by digestive enzymes (McClements, Decker, & Park, 2009). This interest can be partially explained because of the nutritional quality of lipids, which provide a high amount of energy in comparison with other nutrients (9 kcal/g) and are source of several bioactive compounds (sterols, vitamins, essential long-chain polyunsaturated fatty acids, etc).
In this review the digestion of food lipids and the techniques used for its monitoring will be discussed. Although several methodologies have been employed in food lipid research, each of them presents different advantages and drawbacks, in such a way that the study of the advance of lipolysis and of the bioaccessibility of lipidic components (release of potentially absorbable molecules in the gastrointestinal lumen) still can be considered as a challenging task.
Lipolysis reaction taking place during digestion
Food lipids mostly consist of triglycerides (TG), which are esters made up from the esterification of three fatty acids (FA) with a molecule of glycerol (Gly). However, as TG cannot be directly absorbed by the intestinal cells, a process of hydrolysis of the ester bonds is required before their incorporation into the bloodstream. For this purpose, several types of lipases (gastric and pancreatic lipases, among them colipase-dependent lipase, carboxyl ester hydrolase or bile salt stimulated lipase and phospholipase A2) are secreted within digestive juices, ensuring the absorption of 95% of ingested lipids in the form of monoglycerides (MG) and FA. Both are the only lipolytic products arising from TG that can be absorbed. In healthy adults, hydrolysis of TG mainly occurs in the first section of the small intestine (duodenum) due to the activity of colipase-dependent lipase at the lipid-water interface (Reis, Holmberg, Watzke, Leser, & Miller, 2009; Golding & Wooster, 2010).
The hydrolysis of TG proceeds as a 2-step reaction, which is directed by the regiospecificity of human lipases. Firstly, the hydrolysis of the ester bond in position 3 of a TG yields a FA and a 1,2-diglyceride (1,2-DG). Secondly, this 1,2-DG is hydrolyzed in position 1 to release a second FA and the corresponding 2-monoglyceride (2-MG) (Desnuelle & Savary, 1963; Mattson & Volpenhein, 1964). Isomerization reactions of 1,2-DG into 1,3-DG and of 2-MG into 1-MG can also occur in the gastrointestinal lumen, making possible the complete hydrolysis of TG into three FA and a molecule of Gly (Miettinen & Siurala, 1971; Borgström, Tryding, & Westöö, 1957; Borgström, 1964).
Therefore, techniques able to identify and quantify all the several kinds of molecular species (including isomers) that may be formed during digestion of TG are needed for a deep study of lipid hydrolysis and for a proper assessment of the advance of lipolysis reaction.
Methodologies usually employed for the study of lipid digestion
As previously commented, several analytical techniques are being used in lipid digestion research, either when performing in vivo, ex vivo or in vitro experiments.
1) Titration of fatty acids
One of the techniques most commonly employed to measure the rate of lipolysis is the titration of released FA. The equipment used for this purpose is named pH-stat titration unit, which records the volume of an alkaline solution (usually NaOH) that is continuously added to the reaction medium in order to maintain the pH at a constant value (Beisson, Tiss, Riviere, & Verger, 2000). In fact, as lipases release FA from the different glycerides (TG, DG, MG), the pH of the medium tends to drop. Then, the molar percentage of FA can be estimated in a very simple way by calculating the number of moles of alkali consumed divided by the number of moles of FA that would arise from TG after being digested. Due to its ease of use, this technique is one of the most commonly employed to estimate the extent of lipid digestion (Fatouros, Bergenstahl, & Mullertz, 2007; Brogård, Troedsson, Thuresson, & Ljusberg-Wahren, 2007; Li & McClements, 2010; Helbig, Silletti, Timmerman, Hamer, & Gruppen, 2012; Lamothe, Corbeil, Turgeon, & Britten, 2012; Marze, Menier, & Anton, 2013; Zhu, Ye, Verrier, & Singh, 2013).
Nevertheless, this methodology presents some limitations. The first drawback is that pH-stat can only be used in in vitro experiments in which lipolysis reactions occur in a closed vessel where conditions can be continuously controlled. Moreover, this methodology is a one-step procedure, generally employed to monitor the activity of pancreatic lipases during the intestinal step. In the case of investigating the activity of gastric lipases, analysis by this methodology becomes more tedious, because a back-titration is also required in order to take into account those FA which are not ionised at low pH values (Beisson et al., 2000).
In addition to this, the quantitative information obtained by means of pH-stat titration technique is very limited because only information regarding FA is obtained, leaving unknown the number of moles of 1- and 2-MG, as well as of 1,2- and 1,3-DG generated during lipolysis. Bearing in mind that MG are also potentially absorbable molecules, pH-stat titration technique offers a very partial view of lipolysis reaction, hampering the proper assessment of lipid bioaccessibility. Several authors even assume in their calculations that complete hydrolysis of TG into three FA and one molecule of Gly does not occur (Pafumi et al., 2002; Li & McClements, 2010; Lamothe et al., 2012; Marze et al., 2013), although it is well known that it does (Borgström, Tryding, & Westöö, 1957; Borgström, 1964; Mattson & Volpenhein, 1964); this inadequate assumption leads to a noticeable overestimation of the advance of lipid digestion.
Furthermore, the results obtained by means of this methodology might not be very accurate. In fact, the drop of pH in the digestion vessel may not always be representative of the FA released, especially when using complex matrices (such as foods) or complex digestion juices (with compositions similar to those of human juices). For instance, the buffering capacity of certain components like proteins, present in either food or digestive juices themselves, can counterbalance the decrease of pH, underestimating the advance of hydrolysis reaction. For this reason, simple solutions whose composition widely differs from in vivo digestion juices are employed as buffers (Di Maio & Carrier, 2011). Besides, the accuracy of the results obtained by titration is also dependent on both the ionization of each FA and its availability to be titrated. Indeed, the lipid composition in the several kinds of acyl groups, the pH of the reaction medium and the concentration of bile salts and electrolytes can greatly influence the volume of alkaline solution to be consumed (Sek, Porter, & Charman 2001; Kanicky & Shah, 2003; Thomas, Holm, Rades, & Müllertz, 2012). For example, the amount of alkali consumed to neutralize 1 mol of butyric acid (C4:0) can be 1000-fold higher than that employed to neutralize 1 mol of stearic acid (C18:0); likewise, the pKa of a mixture of several FA might differ from the pKa of the single FA (Zhu et al. 2013; Kanicky & Shah, 2003). Thus, in certain cases, the moles of alkali calculated might not be equivalent to those of the FA released.
Finally, the selection of the end point value of the pH is of primary importance. This latter should be higher than the apparent pKa of the mixture of FA in order to ensure that all the carboxylic groups of FA are in their ionized form, as well as to increase their solubility in water and thus, their availability for neutralization. At pH ranging from 9 to 10, the ionic repulsion between adjacent ionized carboxylic groups is increased, enhancing the solubility of FA (Kanicky & Shah, 2003). Not only would the presence of FA negatively charged and of FA dimmers negatively affect the reliability of titration, but also the potential formation of calcium soaps. Indeed, the complexation of FA with cations would also decrease the volume of alkali consumed, the number of moles of FA released and thus, the advance of hydrolysis reaction could be underestimated. In this regard, the influence of the compositions of digestive juices and of food matrices is considered of paramount importance.
2) Chromatographic techniques
Apart from pH-stat titration, several studies have employed chromatographic techniques to assess the extent of lipid digestion. Among these can be cited High Performance Liquid Chromatography coupled to an Evaporative Light Scattering Detector (HPLC-ELSD) (Martin, Nieto-Fuentes, Señoráns, Reglero, & Soler-Rivas, 2010; Kenmogne-Domguia, Meynier, Viau, Llamas, & Genot, 2012), Thin Layer Cromatography coupled to Flame Ionisation Detector (TLC-FID) (Capolino et al., 2011) or to video densitometry (Armand et al., 1999; Sek et al., 2001), Gas Chromatography followed by Mass Spectrometry (GC-MS) (Shen, Apriani, Weerakkody, Sanguansri, & Augustin, 2011; Ye, Cui, Zhu, & Singh, 2013; Zhu et al., 2013) or by FID (GC-FID) (Helbig et al., 2012). Other less common methodologies, such as Ultra High Liquid Chromatography-Electrospray Ionization/ Mass Spectrometry (UHPLC-ESI/MS), have been also employed for the study of lipolysis advance during in vitro digestion (Tarvainen, Suomela, & Kallio, 2011; Tarvainen, Phuphusit, Suomela, Kuksis, & Kallio, 2013). By means of the above-mentioned methodologies, separation, identification and quantification of the different molecular species that may be generated during the hydrolysis of TG can be carried out.
However, it has to be taken into account that several preparation steps are often needed prior to analysis in order to chemically modify the sample. For example, the estimation of the content of FA in the digested lipid extract by means of GC-MS requires both an acid and an alkaline esterification of the FA and/or transesterification of acyl groups present in the sample and further quantification of the methyl esters obtained. In acid medium FA and acyl groups present in the several kinds of glycerides (TG, DG and MG) are (trans)esterified, whereas in the alkaline one, only the acyl groups are transesterified. Afterwards, the methyl esters obtained (commonly known as Fatty Acid Methyl Esters or FAMEs) are quantified and the content of FA in a digested sample is estimated by difference (Shen et al., 2011; Zhu et al., 2013). Thus, these multi-step chromatographic techniques are quite tedious and laborious.
Likewise, when using these methodologies, reference compounds and calibration curves are needed for quantification purposes. For example, in the case of analysis by means of HPLC-ELSD, TLC-FID or TLC coupled to video densitometry, after separation of TG, MG, DG and FA by chromatography, identification is performed by comparison of retention times with that of pure standard compounds. Then, calibration curves of each of the standard compounds are needed for further quantification.
In comparison with the pH-stat titration method, these methodologies can also be considered less environmentally friendly because of the large amounts of polluting organic solvents used.
Furthermore, taking into account that certain discrepancies among data obtained with the above-mentioned methodologies have been reported (Sek et al., 2001; Helbig et al., 2012; Thomas et al., 2012), sound methodological developments are still needed.
3) Nuclear Magnetic Resonance
Spectroscopic techniques, such as Nuclear Magnetic Resonance (NMR), have been previously applied to identify and quantify DG, MG and FA in fats, oils or other lipidic mixtures. Quantification by NMR is based on the premise that the signal produced by exciting a nucleus from a fully relaxed state is directly proportional to the number of molecules containing the nucleus of interest (Fernandes, de Souza, & de Vasconcellos Azeredo, 2012).
With regard to 13C NMR, signals associated with the glycerol carbon atoms and the first two carbon atoms in the acyl chains have been used to identify and quantify partial glycerides in standard mixtures and naturally present in complex mixtures of several glycerol esters and oils (Gunstone, 1991; Fernandes et al., 2012; Vlahov, 1996, 2006). Although the different isomers of DG and MG can be differentiated, the main disadvantage of this technique is that it involves long relaxation delays and lengthy accumulations to achieve a satisfactory signal to noise ratio necessary for an accurate quantification. Moreover, the use of internal and external reference compounds is also required for calibration curves.
Regarding 31P NMR spectroscopy, Spyros & Dais (2000) developed a methodology to determine the content of MG and DG in vegetable oils. This technique showed an excellent resolution between the chemical shifts of the phosphorylated hydroxyl groups present in 1-MG, 2-MG, 1,2-DG and 1,3-DG, allowing a reliable quantification of these lipolytic products. Nevertheless, a previous derivatization of the labile hydrogens in partial glycerides with 2-chloro-4,4,5,5-tetramethyldioxaphospholane is required, as well as the introduction of cyclohexanol as internal standard in the reaction mixture for the subsequent quantification of the phosphorylated derivatives.
As far as 1H NMR is concerned, a more extensive overlapping of the spectral signals comparing to 13C NMR and 31P NMR occurs, because of the shorter range of chemical shifts. Nevertheless, in spite of the above-mentioned disadvantage, recent studies have demonstrated the usefulness of this technique to study in detail lipolysis reaction occurring during digestion (Nieva-Echevarría, Goicoechea, Manzanos, & Guillén, 2014, 2015). In fact, the 1H NMR spectra of TG, 1,2-DG, 1,3-DG, 2-MG, 1-MG and FA greatly differ, especially regarding the spectral region ranging from 3.50 to 5.30 ppm, where specific signals related to the protons in the glycerol backbone of glycerides appear, and the spectral region ranging from 2.25 to 2.45 ppm, where protons of methylenic groups in α-position in relation to the carbonyl group of FA and all acyl groups are visible (except those of docosahexaenoic acid/acyl group). Differences among the 1H NMR spectra of TG, partial glycerides and FA can be observed in Figure 1.
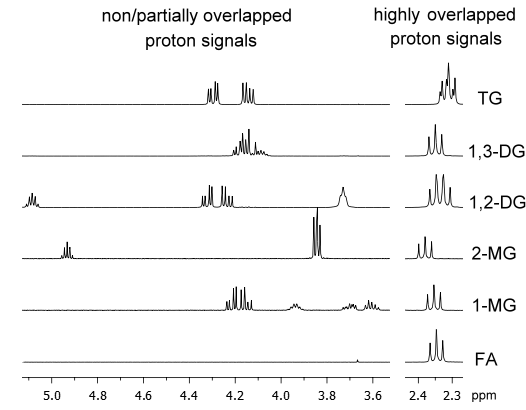
Figure 1. Enlargements of certain spectral regions of the 1H NMR spectra of triolein, 1,3- and 1,2-diolein, 2- and 1-monoolein and oleic acid.
Since most of these 1H NMR signals do not overlap or only do so partially, the identification of the different glycerides present in a lipid hydrolysate can be easily performed by the simple observation of the presence/absence of the corresponding signals in the spectrum. Thus, no chemical modification of the lipid sample is needed.
In addition, due to the proportionality existing between the area of the 1H NMR spectral signals and the number of protons that generate them, quantitative information on the proportions of the several kinds of lipolytic products can be easily obtained just by applying different equations and calculating the intensity of specific spectral signals. In this case, the performance of calibration curves with standards for each one of the compounds under study is not required. The accuracy of the results obtained with 1H NMR spectral data was validated by using mixtures of known composition made up with several standard compounds which simulated lipid hydrolysates from different origins (vegetable or animal). Comparison of the molar percentages of TG, DG, MG and FA obtained by weight and those obtained by applying the new developed equations showed a very high level of agreement, the error in the determination ranging from 0 to 9% (Nieva-Echevarría, Goicoechea, Manzanos, & Guillén, 2014).
In later studies, the application of this new approach to study qualitatively and quantitatively the changes due to the progression of lipolysis under digestive conditions was carried out (Nieva-Echevarría, Goicoechea, Manzanos, & Guillén, 2015, 2016, 2017).
As shown in Figure 2, significant changes in the 1H NMR spectra of lipids occur as lipolysis advances: spectral signals corresponding to TG tend to disappear, whereas other new signals (related with lipolytic products) appear and show higher intensity as digestion advances. Hence, by means of this technique, it is possible to discriminate among samples with different lipolytic levels by the simple observation of their spectra, in a rapid way and without further enlargement.
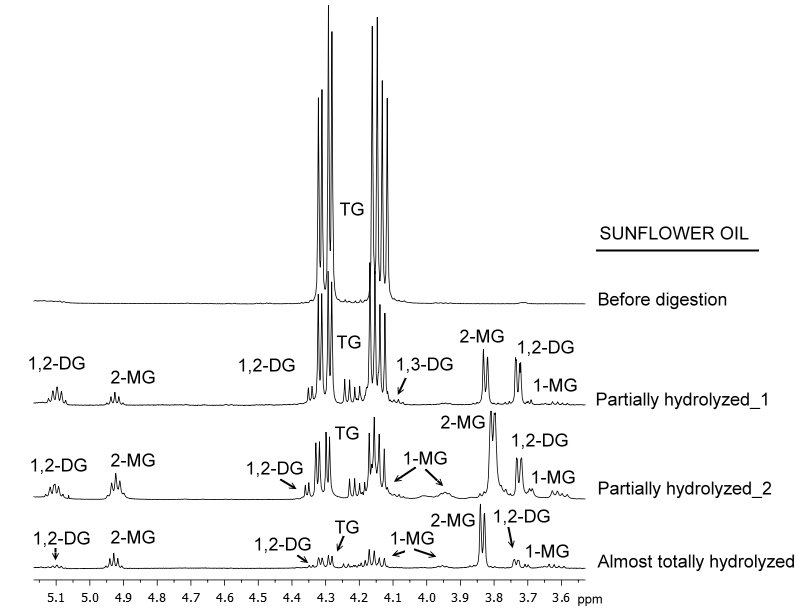
Figure 2. Enlargements of certain spectral regions of the 1H NMR spectra of sunflower oil before and during in vitro digestion process.
In addition, the high versatility of 1H NMR enables the assessment of the extent of lipolysis reaction in any of its current meanings (lipid bioaccessibility, percentage of FA physiologically releasable, hydrolysis level or degree of TG transformation), in contrast to chromatographic and pH-stat titration techniques (Nieva-Echevarría et al., 2015).
In summary, 1H NMR allows a global qualitative and quantitative study of lipid digestion in a simple and fast way, and without any chemical modification of the sample. It must be noted that the advantages of this technique for the evaluation of lipolysis degree should not only be considered in the fields of food technology and nutrition, but also in those of enzymology, pharmacology, medicine and petrochemistry, among others.
This work has been supported by the Spanish Ministry of Economy and Competitiveness (MINECO, AGL2015-65450-R), by the Basque Government (EJ-GV, GIC10/85-IT-463-10 and PA18/04) and by the Unit for Education and Research “Food Quality and Safety” (UPV/EHU-UFI-11/21).
Armand M, Pasquier B, André M, Borel P, Senft M, Peyrot J, Salducci J, Portugal H, Jaussan V, Lairon D (1999) Digestion and absorption of 2 fat emulsions with different droplet sizes in the human digestive tract. Am J Clin Nutr 70(6): 1096-1106. PMid:10584056
View Article PubMed/NCBIBeisson F, Tiss A, Riviere C, Verger R (2000) Methods for lipase detection and assay: a critical review. Eur J Lipid Sci Technol 102(2): 133-153. 1438-9312(200002)102:2<133::AID-EJLT133>3.0.CO;2-X
View ArticleBorgström B, Tryding N, Westöö G (1957) On the extent of hydrolysis of triglyceride ester bonds in the lumen of human small intestine during digestion. Acta Physiol Scand 40(2-3): 241-247. PMid:13469530
View Article PubMed/NCBIBorgström B (1964) Influence of bile salt, pH, and time on the action of pancreatic lipase; physiological implications. J Lipid Res 5: 522-531.
Brogård M, Troedsson E, Thuresson K, Ljusberg-Wahren H (2007) A new standardized lipolysis approach for characterization of emulsions and dispersions. J Colloid Interface Sci 308(2): 500-507. PMid:17289070
View Article PubMed/NCBICapolino P, Guérin C, Paume J, Giallo J, Ballester JM, Cavalier J F, Carrière F (2011) In vitro gastrointestinal lipolysis: Replacement of human digestive lipases by a combination of rabbit gastric and porcine pancreatic extracts. Food Dig 2(1-3): 43-51.
View ArticleDesnuelle P, Savary P (1963) Specificity of lipases. J Lipid Res 4(4): 369-384. PMid:14168179
PubMed/NCBIDi Maio S, Carrier R L (2011) Gastrointestinal contents in fasted states and post-lipid ingestion. In vivo measurements and in vitro models for studying oral drug delivery. J Control Release 151(2): 110-122. PMid:21134406
View Article PubMed/NCBIFatouros DG, Bergenstahl B, Mullertz A (2007) Morphological observations on a lipid-based drug delivery system during in vitro digestion. Eur J Pharm Sci 31(2): 85-94. PMid:17418543
View Article PubMed/NCBIFernandes JLN, de Souza ROMA, de Vasconcellos Azeredo RB (2012) 13C NMR quantification of mono and diacylglycerols obtained through the solvent-free lipase-catalyzed esterification of saturated fatty acids. Magn Reson Chem 50(6): 424-428. PMid:22539418
View Article PubMed/NCBIGolding M, Wooster TJ (2010) The influence of emulsion structure and stability on lipid digestion. Curr Opin Colloid Interface Sci 15(1–2): 90-101.
View ArticleGunstone FD (1991) 13C-NMR studies of mono-, di-and tri-acylglycerols leading to qualitative and semiquantitative information about mixtures of these glycerol esters. Chem Phys Lipids 58(3): 219-224. 90095-S
View ArticleHelbig A, Silletti E, Timmerman E, Hamer RJ, Gruppen H (2012) In vitro study of intestinal lipolysis using pH-stat and gas chromatography. Food Hydrocolloids 28(1): 10-19.
View ArticleKanicky JR, Shah DO (2003) Effect of premicellar aggregation on the pKa of fatty acid soap solution. Langmuir 19: 2034-2038.
View ArticleKenmogne-Domguia HB, Meynier A, Viau M, Llamas G, Genot C (2012) Gastric conditions control both the evolution of the organization of protein-stabilized emulsions and the kinetic of lipolysis during in vitro digestion. Food Funct 3(12): 1302-1309. PMid:22918290
View Article PubMed/NCBILamothe S, Corbeil MM, Turgeon SL, Britten M (2012) Influence of cheese matrix on lipid digestion in a simulated gastro-intestinal environment. Food Funct 3(7): 724-731. PMid:22476332
View Article PubMed/NCBILi Y, McClements DJ (2010) New mathematical model for interpreting pH-stat digestion profiles: Impact of lipid droplet characteristics on in vitro digestibility. J Agric Food Chem 58(13): 8085-8092. PMid:20557040
View Article PubMed/NCBIMartin D, Nieto-Fuentes JA, Se-oráns FJ, Reglero G, Soler-Rivas C (2010) Intestinal digestion of fish oils and ω-3 concentrates under in vitro conditions. Eur J Lipid Sci Technol 112(12): 1315–1322.
View ArticleMarze S, Meynier A, Anton M (2013) In vitro digestion of fish oils rich in n-3 polyunsaturated fatty acids studied in emulsion and at the oil-water interface. Food Funct 4(2): 231-239. PMid:23086175
View Article PubMed/NCBIMattson FH, Volpenhein RA (1964) The digestion and absorption of triglycerides. J Biol Chem 239(9): 2772-2777. PMid:14216426
PubMed/NCBIMcClements DJ, Decker EA, Park Y (2009) Controlling lipid bioavailability through physicochemical and structural approaches. C Rev Food Sci Nutr 49(1): 48-67. PMid:18949598
View Article PubMed/NCBIMiettinen TA, Siurala M (1971) Bile salts, sterols, sterol esters, glycerides and fatty acids in micellar and oil phases of intestinal contents during fat digestion in man. J Clin Chem Clin Biochem 9(1): 47-52.
View ArticleNieva-Echevarría B, Goicoechea E, Manzanos MJ, Guillén MD (2014) A method based on 1H NMR spectral data useful to evaluate the hydrolysis level in complex lipid mixtures. Food Res Int 66: 379-387.
View ArticleNieva-Echevarría B, Goicoechea E, Manzanos MJ, Guillén MD (2015) Usefulness of 1H NMR in assessing the extent of lipid digestion. Food Chem 179: 182-190. PMid:25722153
View Article PubMed/NCBINieva-Echevarría B, Goicoechea E, Manzanos MJ, Guillén MD (2016) A study by 1H NMR on the influence of some factors affecting lipid in vitro digestion. Food Chem 211: 17-26. PMid:27283602
View Article PubMed/NCBINieva-Echevarría B, Goicoechea E, Manzanos MJ, Guillén MD (2017) 1H NMR and SPME-GC/MS study of hydrolysis, oxidation and other reactions occurring during in vitro digestion of non-oxidized and oxidized sunflower oil. Formation of hydroxy-octadecadienoates. Food Res Int 91: 171-182. PMid:28290321
View Article PubMed/NCBIPafumi Y, Lairon D, Porte PL, Juhel C, Storch J, Hamosh M, Armand M (2002) Mechanisms of inhibition of triacylglycerol hydrolysis by human gastric lipase. J Biol Chem 277(31): 28070-28079. PMid:11940604
View Article PubMed/NCBIReis P, Holmberg K, Watzke H, Leser ME, Miller R (2009) Lipases at interfaces: A review. Adv Col Interface Sci 147–148: 237-250. PMid:18691682
View Article PubMed/NCBISek L, Porter CJH, Charman WN (2001) Characterisation and quantification of medium chain and long chain triglycerides and their in vitro digestion products, by HPTLC coupled with in situ densitometric analysis. J Pharm Biomed Anal 25(3–4): 651-661. 00528-8
View ArticleShen Z, Apriani C, Weerakkody R, Sanguansri L, Augustin MA (2011) Food matrix effects on in vitro digestion of microencapsulated tuna oil powder. J Agric Chem 59(15): 8442-8449. PMid:21721584
View Article PubMed/NCBISpyros A, Dais P (2000) Application of 31P NMR spectroscopy in food analysis. 1. Quantitative determination of the mono- and diglyceride composition of olive oils. J Agric Food Chem 48(3): 802-805. PMid:10725153
View Article PubMed/NCBITarvainen M, Suomela J-P, Kallio H (2011) Ultra high performance liquid chromatography–mass spectrometric analysis of oxidized free fatty acids and acylglycerols. Eur J Lipid Sci Technol 113: 409-422.
View ArticleTarvainen M, Phuphusit A, Suomela J-P, Kuksis A, Kallio H (2013) Effects of Antioxidants on Rapeseed Oil Oxidation in an Artificial Digestion Model Analyzed by UHPLC–ESI–MS. J Agric Food Chem 60(14):3564-3579. PMid:22433015
View Article PubMed/NCBIThomas N, Holm R, Rades T, Müllertz A (2012) Characterising lipid lipolysis and its implication in lipid-based formulation development. AAPS 14(4): 860-871. PMid:22956477
View Article PubMed/NCBIVlahov G (1996) Improved quantitative 13C nuclear magnetic resonance criteria for determination of grades of virgin olive oils. The normal ranges for diglycerides in olive oil. J Am Oil Chem Soc 73(9): 1201-1203.
View ArticleVlahov G (2006) 13C Nuclear Magnetic Resonance. Studies of mono-, di- and triacylglycerols. NOE factors and spin-lattice relaxation of acyl chain carboxy carbons. Triglycerides and cholesterol research. Linda T. Welson (Ed). Nova Publishers Inc, New York.
Ye A, Cui J, Zhu X, Singh H (2013) Effect of calcium on the kinetics of free fatty acid release during in vitro lipid digestion in model emulsions. Food Chem 139: 681-688. PMid:23561161
View Article PubMed/NCBIZhu X, Ye A, Verrier T, Singh H (2013) Free fatty acid profiles of emulsified lipids during in vitro digestion with pancreatic lipase. Food Chem 139(1-4): 398-404. PMid:23561123
View Article PubMed/NCBI