Otávio Augusto Chaves
E-MAIL: otavio_ufrrj@hotmail.com
Molecular Docking and Semi-Empirical Study of the Binding between Allura Red, a synthetic food dye, with Bovine Serum Albumin
Corresponding Author
Affiliation
Otávio Augusto Chaves1,2*, Robson Pacheco Pereira2
1 Chemistry Department, Universidade Federal Rural do Rio de Janeiro (UFRRJ), Rodovia BR-465, Km 7, 23970-000, Seropédica-RJ, Brazil.
2 Instituto Militar de Engenharia (IME), Praça General Tibúrcio 80, 22290-270, Urca, Rio de Janeiro-RJ, Brazil
Article Reviewed By:
Dong-Qiang Lin(lindq@zju.edu.cn)
Balamurali M M(balamurali.mm@vit.ac.in)
Luca Dellafiora(luca.dellafiora@unipr.it)
Fei Zhang(hxxyzf@mail.tjnu.edu.cn)
Citation
Ot
Abstract
The interaction between Bovine Serum Albumin (BSA), one of the standard proteins used to study the bioavailability of biological molecules in the bloodstream, and allura red, a commercial food additive, was studied by molecular docking. The computational results suggest GoldScore as the main function to study the BSA:Allura red interactions. The most probable binding site to Allura red is the site IIA (Sudlow’s site I), where Trp212 residue can be found and the Student's t-distribution indicate that there are statistically significant differences between the two data sets obtained by molecular docking. The molecular docking results suggest that Allura red interacts with Arg-194, Arg-198, Trp-212, Arg-217, Gln-220, Lys-294 and Ser-343 residues, by different intermolecular interactions (electrostatic forces, hydrogen bonding and hydrophobic interactions). After semi-empirical optimization, the amino acid residues Gln-220 and Lys-294 exhibited a significantly different interaction structure with Allura red. Theoretical interaction enthalpy change value (△Hint=14.0 kJ/mol) reinforces the proposal that the preferential interaction cavity for Allura red in BSA is the Trp-212containing site.
Introduction
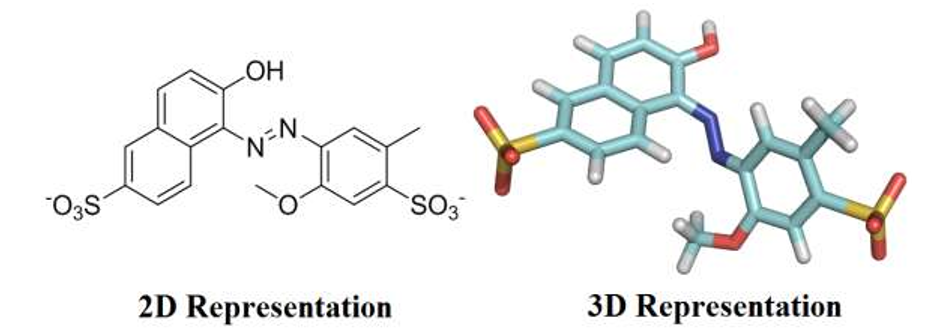
Food dyes are complex organic chemicals that were originally derived from coal tar, but are currently obtained from petroleum. Dyes are largely used in food industry to improve the appearance of products aiming to attract consumers by visual appeal. Compared with natural dyes, industrial food producers use synthetic colorants due to their high stabilities under light, oxygen and pH, color uniformity, low microbiological contamination and relatively lower production costs [1]. However, some synthetic colorants raise significant health concerns to human body, particularly if they are excessively consumed. Over the past century, more food dyes have been found to be riskier than any other food additive categories, therefore, controlling of dyes dosage is of considerable importance in food industry [2,3]. Red 40, also known as Allura Red (Figure 1), is a water-soluble monoazo colorant, approved by Food and Drug Administration (FDA) for use in beverages, bakery goods, dessert powders, candies, cereals, foods, drugs, and cosmetics and, in terms of pounds consumed, is by far the most used red dye. Red 40 has an Acceptable Daily Intake (ADI) of 7 mg/kg bw/day [4]. That ADI translates into 210 mg for a 30-kg child. Companies produce the equivalent of about 25 mg of the dye per person per day, with many children, to whom colorful cereals, candies, snack foods, and dairy products are marketed, may consume several times as much. However, if excessively consumed, Allura Red has been related to allergies, toxic effects, and hyperactivity in children [5].
Serum albumins (bovine serum albumin – BSA – and human serum albumin – HSA) are the major soluble protein constituents of the circulatory system and have many physiological functions. The most important property of this group of proteins is that they serve as a depot protein and as a transport protein for many drugs and other bioactive small molecules e.g. fatty acids, bilirubin, prostaglandins, steroids, cholesterol, uric acid, hormones, vitamins, metal ions, drugs and contrasting substances [6,7]. BSA has been one of the most extensively studied of this group of proteins, due to its availability, lower cost and particularly due to its structural homology with HSA (76% identity and 88% similarity) [8]. BSA is a single-chain protein of 582 amino acids with two tryptophan residues (Trp-134 and Trp-212). The primary structure of BSA consists in three domains (I, II and III) and each one consists of two sub-domains (A and B) [9]. Extensive investigations into the interaction between serum albumin and natural or synthetic compounds showing biological activity have been reported in the literature [10-15].
Since serum albumin serves as a transport carrier for molecules and plays a dominant role on their bioavailability, the binding of food additives such as food dyes on serum albumins may dramatically affect the structure and transport functions of these proteins, as well as changing the metabolism, distribution, and excretion of these molecular species [16,17]. Therefore, the investigation of interactions between food colorants and serum albumin is important for food chemistry, life sciences, and clinical medicine. In the literature, the interaction between HSA:Allura red was investigated by spectroscopic techniques and computational methods [2]. On the other hand, the interaction between BSA:Allura red was investigated by spectroscopic techniques [17]. In both serum albumins the main fluorescence quenching mechanism is static, indicating a complex formation in the ground state. There is one main binding site in serum albumins to allura red (Sudlow’s site I) and the binding affinity is moderate, with a favorable enthalpy change value. The main difference in the association of allura red with the serum albumins is the negative enthalpy change value for HSA:Allura red and positive enthalpy change value for BSA:Allura red, suggesting different intermolecular interactions between the amino acid residues with the ligand in the complex stabilization [2,17]. In order to provide a molecular level explanation for the BSA:Allura red binding, aiming to estimate the participation of specific chemical groups and their interactions in complex stabilization, a molecular docking exercise was carried out. This theoretical study provides relevant aspects for proper and comprehensive understanding on the interaction of Allura red, a commercial food dye, with BSA.
Materials & Methods
2.1. Methodology for molecular docking studies
The crystallographic structure of bovine serum albumin was obtained from the Protein Data Bank (PDB) with access code 4F5S [18]. This structure has a resolution of 2.47 Å. The Allura red structure was built and energy-minimized with the Density Functional Theory (DFT), method Becke-3-Lee Yang Parr (B3LYP) with the standard 6-31G* basis set, available at the Spartan'14 program (Wavefunction, Inc.).
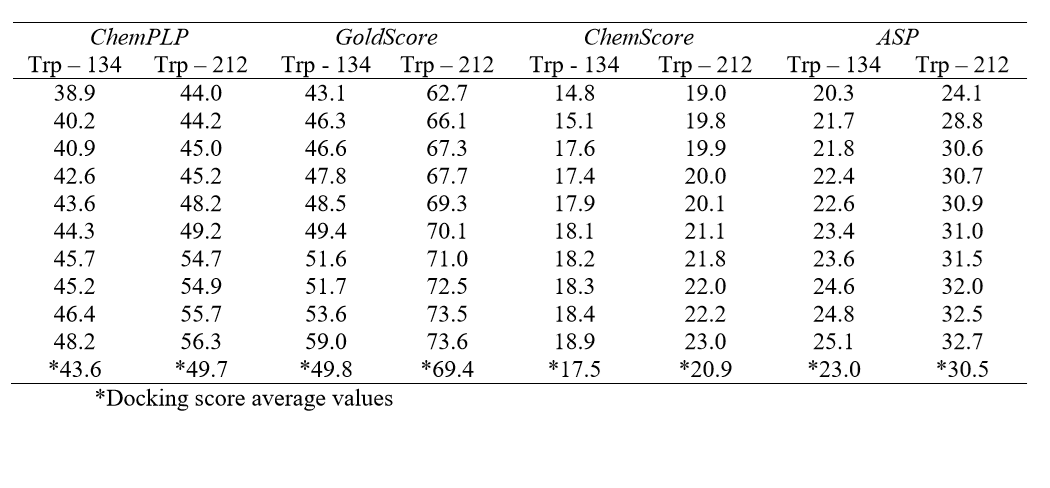
Molecular docking was performed with the GOLD 5.2 software (CCDC). Hydrogen atoms were added to the protein according to the data inferred by the program on the ionization and tautomeric states [19]. From the literature, the main binding sites of BSA for different classes of molecules known are: Trp-134-containing binding site (subdomain IB) and Trp-212-containing binding site (subdomain IIA) [17]. In order to identify the main binding site on the protein, a spherical domain of 10 Å radius around each tryptophan residue was defined, then docking calculations with the ligand (Allura red) inside each of those domains were carried out, considered as the two possible interaction cavities. The score of each pose identified was calculated as the negative of the sum of a series of energy terms involved in the protein-ligand interaction process, so that the more positive the score, stronger is the interaction [20]. The number of genetic operations (crossover, migration, mutation) in each docking run used in the search procedure was set to 100,000. The software optimizes hydrogen-bond geometries by rotating all hydroxyl and amino groups of the amino acid side chains. The scoring functions used were ChemPLP, GoldScore, ChemScore and Astex Statistical Potential (ASP), however the best results were obtained with GoldScore function. The Student's ttest was used to determine if the difference between any two data sets (Trp-134- and Trp-212-containing binding site) were statistically significant. The figures representing the docking poses to the highest docking score value were generated with the PyMOL Delano Scientific LLC program.
2.2. Methodology for semi-empirical studies
Molecular docking method is commonly used for predicting binding structures of ligands to proteins, however the docking scores are not comparable to experimental data. In order to increase the accuracy for the best molecular docking geometry, and predict the theoretical interaction enthalpy change value (ΔHint), semi-empirical molecular orbital method calculations were performed [21]. First, for the highest-ranked BSA:Allura red docking complex, all residues located into a 5 Å radius sphere around the ligand were selected, using the DeepView-Swiss-PdbViewer 4.1 software (Swiss Institute of Bioinformatics, Lausanne, Switzerland). The selected Trp-212-containing binding site was composed by 23 amino acid residues: Arg-194; Leu-197, Arg-198, Trp-212, Ala-216, Arg-217, Gln-220, Lys-221, Ala-290, Lys-294, Pro-338, Glu-339, Tyr-340, Ala-341, Val-342, Ser-343, Glu-443, Pro-446, Cys-447, Glu-449, Asp-450, Ser-453 and Leu-454. The total charge values of BSA:Allura red complex, Trp-212containing binding site and Allura red, are -1, +1 and -2, respectively.
The structures were subsequently optimized with the semi-empirical molecular orbital PM7 method, which is available for the MOPAC2012™ software (Molecular Orbital PACkage, Stewart Computational Chemistry, Colorado Springs, CO, USA). Considering initially that the ligand and the selected binding pocket are involved in an aqueous phase and when the BSA:Allura red complex is formed the hydrophobicity inside the binding pocket increases, as a result of the positive experimental entropy change value [2,17], the best approximation to the dielectric constant (ε) of the continuum model for Allura red and Trp-212-containing binding site is 78.4 and for BSA:Allura red complex is 4.0 [20]. After a previous geometry optimization of all hydrogen atoms of each structure, the main amino acid chain was frozen except to the side chains and to ligand structure. The final geometries were completely optimized to obtain the theoretical enthalpy values for the BSA:Allura red complex, Trp-212containing binding site and Allura red. In order to obtain the theoretical interaction enthalpy change value (ΔHint), the difference between the complex enthalpy and the sum of BSA empty site and Allura red enthalpy values was calculated.
Results
The HSA:Allura red interaction was previously reported in the literature and has a moderate affinity (binding constant - Ka≈104 L/mol), with a static fluorescence quenching mechanism (kq≈1012 L/mol.s), that suggests a complex formation in the ground state. The number of binding sites (n) is approximately 1, indicating only one main binding site in HSA structure for Allura red [2]. Comparing the interaction between Allura red with the other serum albumin (BSA), also the main fluorescence quenching mechanism is static and the binding constant values (Ka≈104 L/mol) show a similar binding affinity, given as the number of binding sites (n≈1) is in total agreement with one main binding site in the BSA structure [17]. In both articles, the authors used specific site markers (warfarin, ibuprofen and digitoxin) to identify the main binding site in the albumins and both results indicate that Allura Red competes with warfarin for the same binding site in serum albumins. This observation suggests that the main binding site for Allura red is the subdomain IIA, named Sudlow’s site I, where Trp-212 residue can be found [2,17]. In order to complement the results of the BSA:Allura red interaction, molecular docking calculations were carried out and are described in the present work.
In the BSA structure, there are two main binding sites: Sudlow’s site I, where the Trp-212 residue is found and Sudlow’s site II, which contains the Trp-134 residue. Table 1 lists all the molecular docking fitness scores for Trp-134 and Trp-212containing binding sites using the ChemPLP, GoldScore, ChemScore and ASP functions. The scale of the score fitness obtained by GoldScore, ChemScore, ASP and ChemPLP functions provide an indication to rank the poses comparatively; more favorable structures (according to each function) exhibit higher scores. The highest docking score average was obtained with GoldScore function, suggesting that this function was able to describe more intermolecular interaction possibilities between the selected protein cavity and Allura red. The GoldScore scoring function (Eq. 1) is a molecular mechanics–like function optimized for prediction of the binding sites of the ligand taking into account four terms, such as energy of hydrogen bonds, van der Waals energy, metal interactions and torsion deformations. The fitness score is taken as the negative of the sum of the component energy terms, so that larger fitness scores are better [22].
Gold Score= Shb_ext+Svdw_ext+Shb_int+Svdw_int (1)
in which, Shb_ext and Shb_int are the protein–ligand hydrogen-bond energy (external Hbond) and intramolecular hydrogen bonds in the ligand, respectively. Svdw_ext and Svdw_int are the protein-ligand van der Waals (vdw) energy (external vdw) and intramolecular strain in the ligand, respectively.
From the docking score average (Table 1), results suggest that Allura red interacts more favorably with BSA into the site containing the Trp-212 residue (docking score average 69.4) than in the site containing the Trp-134 residue (docking score average 49.8). Since a t-test is any statistical hypothesis test in which the test statistic follows a Student's t-distribution under the null hypothesis, it can be used to determine if two sets of data are significantly different from each other. In order to give a statistically significant difference for the theoretical results, a Student's t-test was performed: as the p value (4.03x10-9) is less than 0.05 (95% confidence interval), the null hypothesis is rejected, therefore there is a statistically significant difference between the two protein binding sites [23], and the molecular docking results suggest only one main binding site in the BSA structure for Allura red.
Sudlow’s site I comprises a large hydrophobic cavity, thus interactions in this site occur mainly via hydrophobic interactions. Generally, ligands that strongly bind to site I are generally dicarboxylic acids and/or bulky heterocyclic molecules with a negative charge containing azo and/or sulfur groups (e.g., phenylbutazone, azapropazone, tolbutamide, bucolome and sulfisoxazole) [24], whose structural characteristics are similar to those in the Allura red structure - Allura Red shows a heterocyclic chemical structure, with three benzene rings that confer hydrophobicity to the molecule; in addition, Allura Red forms an anionic compound promoted by both sulfonic groups ionization.
The interactions between endogenous or exogenous agents with proteins (van der Waals and electrostatic forces, including hydrogen bonds and hydrophobic effects) can be related to thermodynamic parameters (enthalpy change values - ΔH°- and entropy change values - ΔS°) [24]. As described in the literature, the thermodynamic parameters (ΔH°=-24.96 kJ/mol and ΔS°=4.96 J/molK) for the HSA:Allura red interaction suggest hydrogen bonding and hydrophobic interactions as the main binding forces in the association [2]. On the other hand, for the BSA:Allura red interaction, the thermodynamic parameters (ΔH°=18.59 kJ/mol and ΔS°=131.8 J/molK) suggest hydrophobic interactions as dominant forces in the system [17]. In order to identify the main binding forces in the association between BSA and Allura red in the Sudlow’s site I, the highest docking score value obtained by GoldScore function was analyzed.
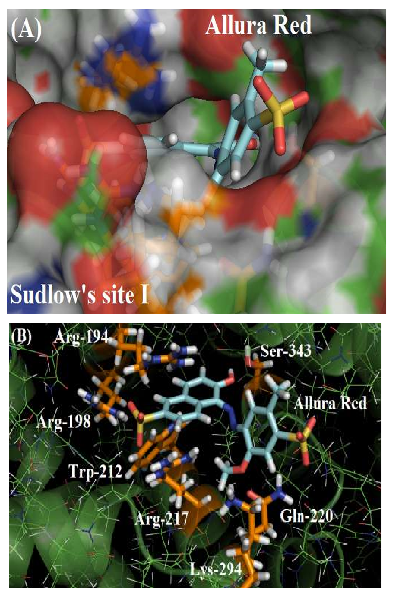
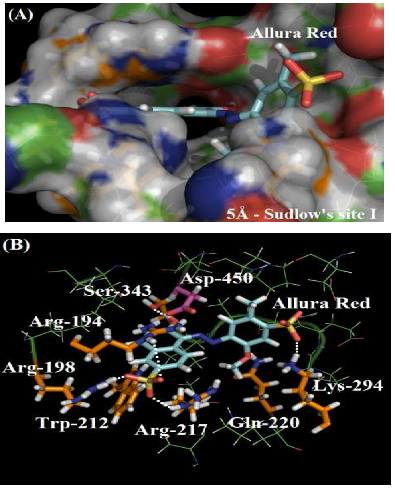
As depicted in Figure 2A, the molecular surface of Sudlow’s site I suggests that one of the benzene rings of Allura red structure is more exposed to the aqueous media instead of located fully inside the protein cavity, justifying the moderate affinity of the dye with the plasmatic albumin. Figure 2B shows the main amino acid residues which interact with Allura red (Arg-194, Arg-198, Trp-212, Arg-217, Gln-220, Lys-294 and Ser-343), by different intermolecular interactions (electrostatic forces, hydrogen bonding and hydrophobic interaction). In the literature, molecular docking studies for HSA:Allura red also suggest the presence of the amino acids arginine, tryptophan, glutamine, lysine and serine residues in the complex stabilization, however the molecular docking studies for BSA:Allura red did not detect the participation of hydrophobic amino acids leucine, phenylalanine, isoleucine, alanine, and valine residues, such as the hydrophilic amino acids cysteine, histidine and glutamic acid residues as described to HSA [2].
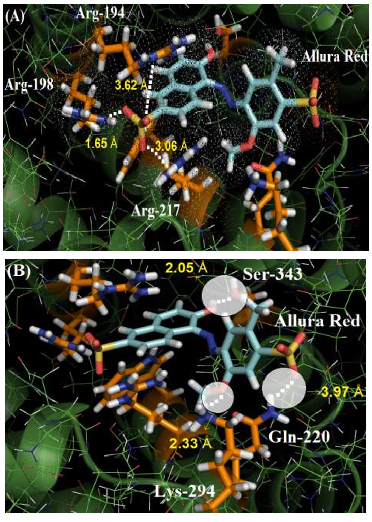
The guanidinium group of the amino acids Arg-194, Arg-198 and Arg-217 residues interact with one of the sulfonic groups of the ligand via electrostatic forces, with distances of 3.62 Å, 1.65 Å and 3.06 Å, respectively. Figure 3A shows the van der Waals surface for each arginine residue and the van der Waals surface for Allura red. The overlaps between the van der Waals surfaces of the selected amino acid residues and the sulfonic group of Allura red indicates that the interaction between these structures is highly possible. On the other hand, Figure 3B suggests the hydrogen bonding formation between the NH of Gln-220 residue (as a donor) with one of the oxygen of the sulfonic group (as an acceptor), with a distance of 3.97 Å, curiously in the literature the same sulfonic group is able to participate in hydrogen bonding to stabilize the complex HSA:Allura red [2]. The molecular docking results also suggest the amino acid residues Lys-294 and Ser-343 can participate in hydrogen bonding with the methoxy and hydroxyl group of the ligand, with distances of 2.33 Å and 2.05 Å, respectively. Finally, the hydrophobic interaction between the Trp-212 residue with the naphthalene ring of the ligand, via t-Stacking, occurs with a distance of 3.80 Å.
The number of amino acid residues in BSA that interacts with Allura red is less than the number of amino acid residues that was previously reported for HSA [2]. This can explain the experimental difference in the enthalpy change values, suggesting that the higher number of amino acid residues in HSA (17 residues) can stabilize more effectively the complex formation (negative enthalpy change value) [2] than the number of amino acid residues (7 residues) in BSA (positive entropy change value) [17].
Semi-empirical calculations were employed to provide a more accurate description of the best molecular docking geometry, and also to predict the theoretical interaction enthalpy change value (ΔHint) in the Trp-212-containing binding site. As depicted in Figure 4A the molecular surface of Sudlow’s site I for the new geometry of the BSA:Allura red complex after semi-empirical optimization suggests that the same benzene ring of Allura red structure is exposed to the aqueous media instead of located fully inside the protein cavity. Figure 4B shows the main amino acid residues which interact with Allura red after semi-empirical optimization (Arg-194, Arg-198, Trp-212, Arg-217, Gln-220, Lys-294 and Ser-343). It should be emphasized that the same amino acid residues, previously described in this work, interact with the ligand, however, with markedly structural differences.
The guanidinium group of amino acid residues Arg-194, Arg-198 and Arg-217 interact with one of the two sulfonic groups of the ligand via electrostatic forces, with distances of 3.68 Å, 1.64 Å and 2.01 Å, respectively. For Arg-194, the increase in the interaction distance is probably due to the competition for the electronic density between sulfonic group of the ligand and the carboxylic group of the Asp-450 residue (represented in pink in Fig. 4B). Ser-343 residue establishes hydrogen bonding interactions with the hydroxyl group of the ligand, with a 1.98 Å distance.
Gln-220 and Lys-294 residues, described by the docking calculations, were able to interact via hydrogen bonding with the sulfonic group and methoxy group of the ligand, respectively, however, in semi-empirical optimized geometry, Gln-220 residue exhibits hydrogen bonding with the methoxy group of the ligand, with a distance of 2.29 Å and Lys-294 residue is interacting favorably with the sulfonic group via electrostatic forces, 1.60 Å apart. Finally, the distance between Trp-212 residue and the naphthalene ring of the ligand decreased, with a distance of 3.40 Å, suggesting a better t-Stacking interaction. Overall, in the semi-empirical description, the amino acid residues Arg-194, Arg-198, Trp-212, Arg-217 and Ser-343 did not exhibit significant differences in the interaction with Allura red, with slightly different distances involved in the intermolecular interaction. On the other hand, the semi-empirical calculations verified a change in the interacting groups in Gln-220 and, for Lys-294 residue, a significant change on the interaction itself.
According to the following representation of the complex formation and Equation 1, the theoretical interaction enthalpy change value (ΔHint) in the Trp-212containing binding site was determined:
BSAcavity + Allura red BSA:Allura red
H int = H BSA: Allura red – (H BSA/cavity + H Allura red) (Eq. 1)
in which HBSA:Allura red is the enthalpy value for the complex (-4575 kJ/mol), HBSA/Cavity is the enthalpy value for the Trp-212-containing binding site (-2673 kJ/mol) and HAllura red is the enthalpy value for the ligand (-1916 kJ/mol). The calculated theoretical interaction enthalpy change was 14.0 kJ/mol. The ΔHint suggests that the binding process is endothermic and the possibility of a direct comparison between experimental interaction enthalpy obtained by Lelis, C.A. et al. 2017 (ΔH°=18.59 kJ/mol) and semi-empirical calculations reinforces the hypothesis that the interaction cavity for Allura red in BSA is the Trp-212-containing site, as well as the main amino acid residues responsible for the BSA:Allura red complex stabilization are those described by theoretical calculations (both molecular docking and semi-empirical calculations). Overall, all the computational results are in total agreement with experimental data previously published for the BSA:Allura red interaction [17].
Conclusion
The computational results suggest GoldScore as the main function for study the BSA:Allura red interaction and the most probable binding site inside the protein structure is the site IIA, known as Sudlow’s site I. Student's t-distribution indicate statistically significant difference between the two data sets (Sudlow’s site I and II) obtained by molecular docking. One of the benzene rings of the Allura red structure is more exposed to the aqueous media instead of being located fully inside the protein cavity, justifying the moderate affinity of the dye with the plasmatic albumin and the molecular docking results suggest that Allura red interacts with Arg-194, Arg-198 and Arg-217 via electrostatic forces, with Trp-212 via hydrophobic interaction, and with Gln-220, Lys-294 and Ser-343 residues via hydrogen bonding. After semi-empirical molecular geometry optimization calculations, the amino acid residues Arg-194, Arg198, Trp-212, Arg-217 and Ser-343 did not exhibit significant differences in their interaction with Allura red, however, for Gln-220 residue, the interacting ligand group was verified to be other than the one predicted by docking calculations and for Lys-294 residue, there was a significant change on the nature of the interaction. Theoretical interaction enthalpy change value (ΔHint=14.0 kJ/mol) obtained in this work is close to the experimental data, reinforcing the hypothesis that the Trp-212-containing site is the most favorable interaction cavity for Allura red in BSA. All computational results are in agreement with experimental data previously reported for the BSA:Allura red interaction by Lelis, C.A. et al. 2017.
Acknowledgement
The authors gratefully acknowledge Prof. Dr. Carlos Mauricio R. Sant’Anna (UFRRJ) for the computational facilities.
Images and Tables